ایمنوپاتولوژی COPD :
ایمنوپاتالوژی این بیماری بر اساس پاسخ این بیماری انطباقی و ذاتی هست این پاسخ به سیگار کشیدن و در معرض دود قرار گرفتن می باشد.
COPD سومین عامل مرگ و میر در دنیا است گرچه یک بیماری قابل درمان و قابل جلوگیری است اما این بیماری با کاهش جریان هوا و که معمولا پیش رونده هستند و همراه هستند با پاسخ التهابی مزمن در راه های هوایی و ریه در پاسخ به ذرات سمی و گازها. حملات این بیماری موجب می شود که شدت بیماری افزایش پیدا کند. علت بیماری هم محیطی است و هم ژنتیک. بنابراین کسانی که سیگاری هستند در حال حاضر عامل 90 درصد از این بیماری ها هستند در حالی که سایر عوامل مانند سوخت های بیومس برای گرمایش و پختن غذا ممکن است عامل مهمی در توسعه این بیماری در شورهای توسعه یافته باشند. تقریبا تنها 25 درصد از سیگاری ها در طول زندگی شان مبتلا به COPD می شوند این می رشاند که عوامل ژنتیک هم در تشکیل این بیماری دخالت دارند تنها عامل ژنتیک شناخته شده کمبود آلفا – ای تی می باشد.
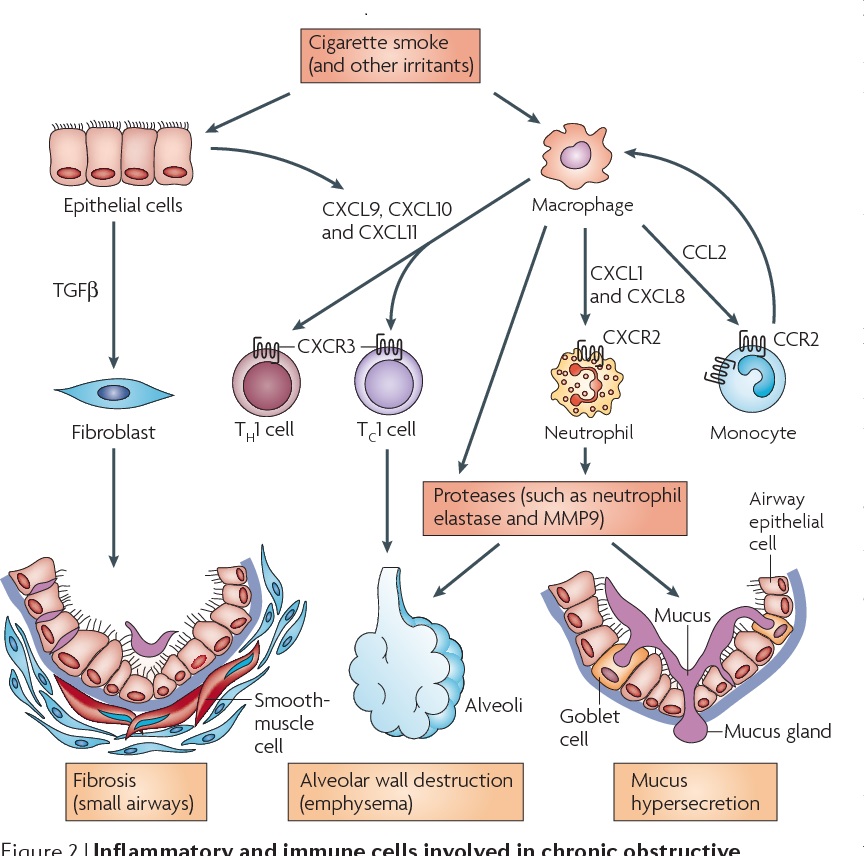
دو پاتالوژی شامل تنگ شدن و تغییر آناتومیک در راه های هوایی کوچک به همراه از دست رفتن و نابودی پارانشیم ریه که در نهایت موجب از دست رفتن اتصال این راه های هوایی از آلبئول ها می شود وجود دارد. این باعث کاهش انقباض پذیری ریه شده و همچنین مقاومت بالا به جریان هوا و بسته شدن راه های کوچک هوایی در حجم بالای ریوی در مدت بازدم می شود. این به نوبه ی خود باعث پرهوایی ریه و احساس تنگی نفس و کاهش ظرفیت حرکتی می شود. هم تغییر آناتومی راه های هوایی کوچک هم تنگی آنها و هم آمپیزم ریوی ناشی از التهاب مزمن اطراف ریه هست.
اکثر مقاومت افزایش پیدا کرده در راه های کوچک تر از دو میلی متر اتفاق می افتد که در بین تقسیم راه های هوایی چهارم تا دوازدهم می باشد. تقریبا 80 درصد از راه های هوایی پشت این تقسیم برونشیول های غیر تنفسی می باشند و بقیه 20 درصد از آنها برونش های کوچک تر هستند که با وجود پلاک های غضروفی در دیواره شان مشخص می شود. پرونشیل ها از برونش ها این تفاوت را دارند که غضروف ندارند و غدد زیر موکوسی هم ندارند.و به طور نسبی سلول های عضلانی صاف بیشتری دارند و سلول های ترشحی موکوس کمتری در لایه اپیتلیال دارند. در افراد طبیعی راه های کوچک به طور جمعی به اندازه یک زمین تنیس می باشد. بنابراین آنها فقط 20 درصد از مقاومت هوایی را تشکیل می دهند. بنابراین 80 درصد از راه های هوایی کوچک باید بسته شوند تا اینکه ما شاهد انسداد راه های هوایی در بسیاری از سیگاری ها باشیم. در نتیجه در بسیاری از سیگاری ها ما شاهد بیماری راه های هوایی کوچک هستیم که متاسفانه توسط سیگاری ها تا زمانی که 80 درصد آنها درگیر نشود احساس نمی شود.
التهاب عامل اصلی بیماری های COPD می باشد. و باعث می شود که راه های هوایی کوچک از نقطه نظر آناتومی تغییر پیدا کند و باعث فعال شدن زیر موکوس که شامل فاصله بین پرده بیس منت تا کناره های سلول های صاف می باشد. مطالعات نشان داده ضایعات راه های هوایی کوچک عامل اصلی پیشروندگی و شدت بیماری COPD است. به همراه آن ما شاهد ارتشاح سلول های التهابی هستند در واقع علت انسداد راه های هوایی در افراد سیگاری ناشی از التهاب راه های هوایی کوچک تغییر آناتومی آنها و از دست رفتن خاصیت انقباض پذیری بافت ریه می باشد اگرچه آنفیزم ریوی با افزایش بیماری همراه می باشد. البته می تواند در بیمارانی که انسداد راه های هوایی ندارند هم اتفاق بیفتد بنابراین آسیب راه های هوایی کوچک شاخصه اصلی شدت و پیشروندگی COPD می باشد و یک تناسب معکوس بین FEV1 و ضخامت راه های هوایی کوچک وجود دارد.
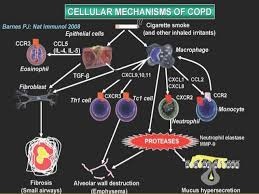
التهاب در بیماران COPD همراه است با افزایش مایکروفاژها نوتروفیل ها و لنفوسیت های بی و تی به اضافه دندرتیک ها. اما وجود نوتروفیل های زیاد و لنفوسیت های بی در اکثر بیماری های شدید دیده می شود. نقش این سلول های التهابی مشخص نیست. در بیماری های COPD متوسط تا ملایم افزایش سلول های التهابی دیده می شود . انتشار ناحیه ای از التهاب در راه های هوایی کوچک وجود دارد در کسانی که سیگار می کشند میزان بیشتری از لوکوسیدها ( CD45+ ) دیده می شود در سابموکوسا. در مطالعات بیشتر نشان داده شد که ضایعات راه های کوچک شاخصه اصلی شدت بیماری COPD است در واقع یک ارتباط معکوس بین ضخامت دیواره ها به کل سطح در راه های کوچک و FEV1 وجود دارد.
توسعه COPD همچنین همراه است با اگزودای موکوسی التهابی در داخل لومن ها و ارتشاح دیوارها توسط پاسخ ایمنی ذاتی و انطباقی که در نهایت فولیکول های لنفوئیدی را تشکیل می دهد. میزان راه های کوچک هوایی که دارای CD4+ و CD8+ و سلول های B فولیکول های لنفوئیدی ماکروفاژها و نوتروفیل ها با شدت COPD همراه است.
اکثر مطالعات مشخص کرده اند که لنفوسیت های سی دی 8 در خون و در بافتهای هوایی پایین ریه در بیماران با شدت ملایم تا متوسط COPD دیده می شود. این سلول ها همچنین در خلط بیماران و لاواژ بیماران دیده می شود اما در این ترشحات تعداد لنفوسیت ها کم است. اما در کسانی که سیگار می کشند و درجاتی از انسداد راه های هوایی دارند و آمفیزم ریوی دارند همه تناسب دارد با افزایش فعالیت CD4+ و CD8+ که فاکتور NF-KAPAB ، استت 4 و اینترفرون گاما و پرفورین افزایش پیدا می کند. نقش CD3+ که لنفسیت تی هست در COPD نا مشخص هست. تعداد سلول های CD8 در خلط در بیماران COPD استیبل نسبت به سیگاری های با عملکرد راه هوایی نرمال یا غیر سیگاری ها افزایش می یابد و این سلول ها فعال بوده و با افزایش سطوح پرفورین همراه است.مقدار CD8 اینترلوکین 4 در خلط در COPD سالم و سیگاری های با عملکرد نرمال ریه و غیر سیگاری ها کاهش می یابد. در حالی که CD8 اینترفرون گاما به شکل زیادی تنها در COPD های کاهش می یابد ارتباط شدیدی بین CD8 اینترلوکین 4 تقسیم بر CD8 اینترفرون گاما و FEV1 فقط در COPD ها دیده می شود.
لنفسیت های تی که اصل آنها CD8 می باشد در موکوس های برونشیال در بیماران COPD دیده می شود اما در بیماران COPD ملایم تا متوسط فرقی در تعداد CD8 – CD3 با افراد غیر سیگاری وجود ندارد.یعنی در افرادسیگاری که مشکل ریوی هنوز پیدا نکرده اند ما افزایش CD3 و CD8 را داریم. در نتیجه تصور می شود که لنفسیت های تی در واقع در اثر سیگار کشیدن بالا می رود.
در COPD ملایم تا متوسط نسبت CD4و CD8 در راه هاهی هوایی کوچک تفاوتی با سیگاری های با راه های هوایی نرمال ندارند.در واقع تعداد سلول های CD4و CD8 در راه های هوایی کوچک با شدت COPD افزایش می یابد. سلول های تی بالغ هرچه بافت های خراب شده بیشتر باشند مقدار آنها هم بیشتر است.
رسپتورهای CCR5 که روی TH1/TC1 دیده می شوند تولید اینترون گاما می کنند در COPD های متوسط و ملایم و در سیگاری های کنترل زیاد می شود. در نتیجه TH1/TC1 نظارتی در COPD ها ملایم تا شدید دیده می شود.
تولید اینترلوکین 4 در غدد بیماران با برونشیت مزمن یک پدیده مستقلی است از لنفسیت های CD8 و در نتیجه ممکن است CD4 در ترشح سایتوکاین های TH2 نقش داشته باشند و در افزایش ترشح در غدد موکوسی در سیگتری ها دیده میشود.
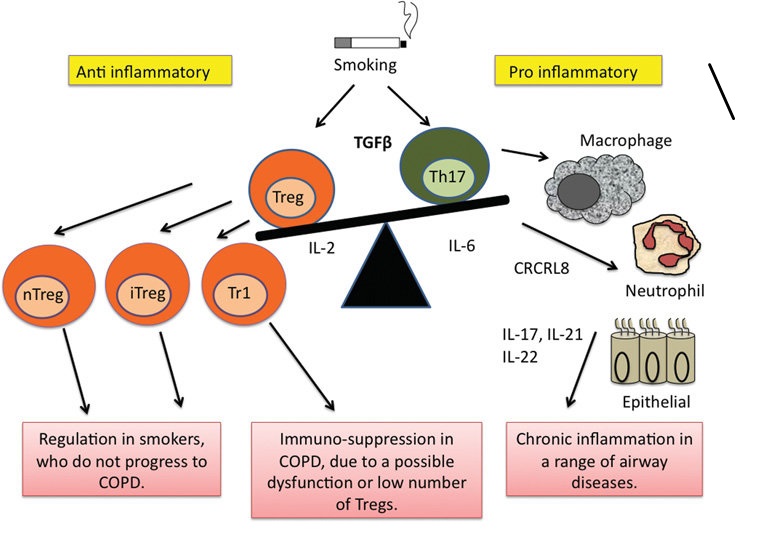
لنفسیت های TH17 موجب ترشح اینتر لوکین 17A,17F می شود که نقش مهمی در تنظیم ارتشاح نوتروفیل ها و ماکروفاژها دارد. و باعث افزایش فعالیت سلول های راه های هوایی تحتانی در COPD می شود. ترشح اینترلوکین 17A,F در اشخاصی که سیگار می کشند زیاد می شود هم در بیماران COPD و هم در بیماران غیر COPD که نشان می دهد ترشح اینترلوکین ها توسط سلول های ریوی می باشد. اینترلوکین 17A در بیماران COPD استیبل دیده می شود.
همچنین سلول های با اکسپرشن اینترلوکین 17A و 22 و سابموکوس های برونشیال بیماران با COPD استیبل زیاد می شود. اگرچه اکسپرشن اینترلوکین 17F و 21 در این گروه ها زیاد نمی شوند. رنگ آمیزی اینترلوکین A,F در سلول های آندوتلیال و التهابی و فیبروبلاست ها دیده می شود. جالب توجه این است که کمتر از 5 درصد از سلول های با اینترلوکین 17+ تی سل هستند و بیشتر از 90 درصد از سلول های اینترلوکین 17A+ در واقع CD31+ هستند که آندوتلیال هستند.به علاوه سلول های اینترلوکین 22 + کمتر از 10 درصدشان تی سل هستند و بیشتر از 80 درصدشان CD31+ هستند که آندو تلیال هستند.
به علاوه تعداد سلول های اینترلوکین 22 و 23 در اپیتلیوم برونشیال در بیماران COPD استیبل زیاد می شود. در تمام سیگاری ها با یا بدون بیماری COPD و در بیماران COPD تنها تعداد سلول های اینترلوکین 22+ متناسب هست با تعداد CD4,CD8+ در موکوس برونشیال
تعداد سلول های التهابی اینترلوکین 17A در زیر اپیتلیوم راه های هوایی کوچک در COPD بیشتر از افراد طبیعی است.اینترلوکین 17A در لنفوسیت ها نوتروفیل ها و ماکروفاژها اکسپرس می شوند. اکسپرسیون اینترلوکین 17F از اینترلوکین 17A در سلول های اپیتلیال و فولیکول های لنفوئیدی بیشتر است. در حالی که اکسپرسیون اینترلوکین 17A در زیر اپیتلیوم از اینترلوکین F بیشتر می باشد.
اکسپرسیون اینترلوکین 17A در COPD های استیبل ولی شدید به طور چشم گیری افزایش می یابد اگرچه CD3+ اینترلوکین 17A را اکسپرس می کند در بیماران COPD خیلی شدید اما اکثر سلول های که اینترلوکین 17A را اکسپرس می کنند ماست سل هستند.CXCL12 به طور چشم گیری در فولیکول های لنفوئیدی در COPD اکسپرس می شوند و این اکسپرسیون در مراحل انتهایی COPD به شدت دیده می شوند این نشان می دهد که اینترلوکین 17A در محیط ریه بیماران شدید و خیلی شدید COPD در پیشرفت بیماری نقش دارند و باعث شکل گیری فولیکول های لنفوئیدی می شوند.
در بیماران COPD تعداد سلول های Tγδ در خلط و لاواژ بیماران COPD به مراتب کمتر از افراد سالم غیر سیگاری است. تعداد این سلول ها به شکل منفی مرتبط است با FEV1 و سال های سیگار کشیدن فقط در گروه COPD .
بررسی سلول های تی CD4 نشان می دهد که میزان مولکول های کنترل کننده TH1 , TH2 و TH17 کاهش پیدا کرده است و این بیماران اسپیرومتری بدی دارند. بررسی CD4 تی سل ها دو فنوتایپ را که با علائم بالینی همراه هستند نشان می دهد. اکسپرسیون اینترلوکین 10 به طور مستقل و معکوسی با آمفیزم مرتبط است ولی با اسپیرومتری هماهنگی ندارد و اکسپرسیون اینترفرون گاما به صورت مستقل و معکوسی همراه است با کاهش اسپیرومتری ولی با کاهش DLCO یا آمفیزم ارتباط ندارد. تحریک سلول های COPD باعث ترشح کمی اینترفرون گاما یا سایر سلول های التهابی می کنند اما بسیاری از بیماران که CL2 و گروه خاطره ای تی افکتورها را کمتر دیده می شود.
نقص CD4+CD25+FOXP3 که سلول های رگولاتوری هستند باعث بیماری ایمنی می شود. تقریبا یک تا سه درصد از CD4TCELL ها TREGS هستند. و در مکان های بافتی تجمع پیدا می کنند که آنتی ژن ها حمله می کنند که در آنجا ایمنی را سرکوب می کنند با ترشح اینترلوکین 10 و TGF-β1 . در حال حاضر اکسپرسیون FOXP3 مارکر مهم ترین مارکر اختصاصی سلول های تی رج هست.
در بیماران COPD استیبل تعداد سلول های CD25++CD45RA+ سلول های تنظیم کننده در حال استراحت و فعال ( CD25++CD45RA- ( در خون کم میشود.
در راههای کوچک COPD میزان CD+CD25+FOXP3 کم میشود که متناسب است با انسداد راههای هوایی. همچنین میزان CD4+CD25+FOXP3 هم در آمفیزم کم میشود.
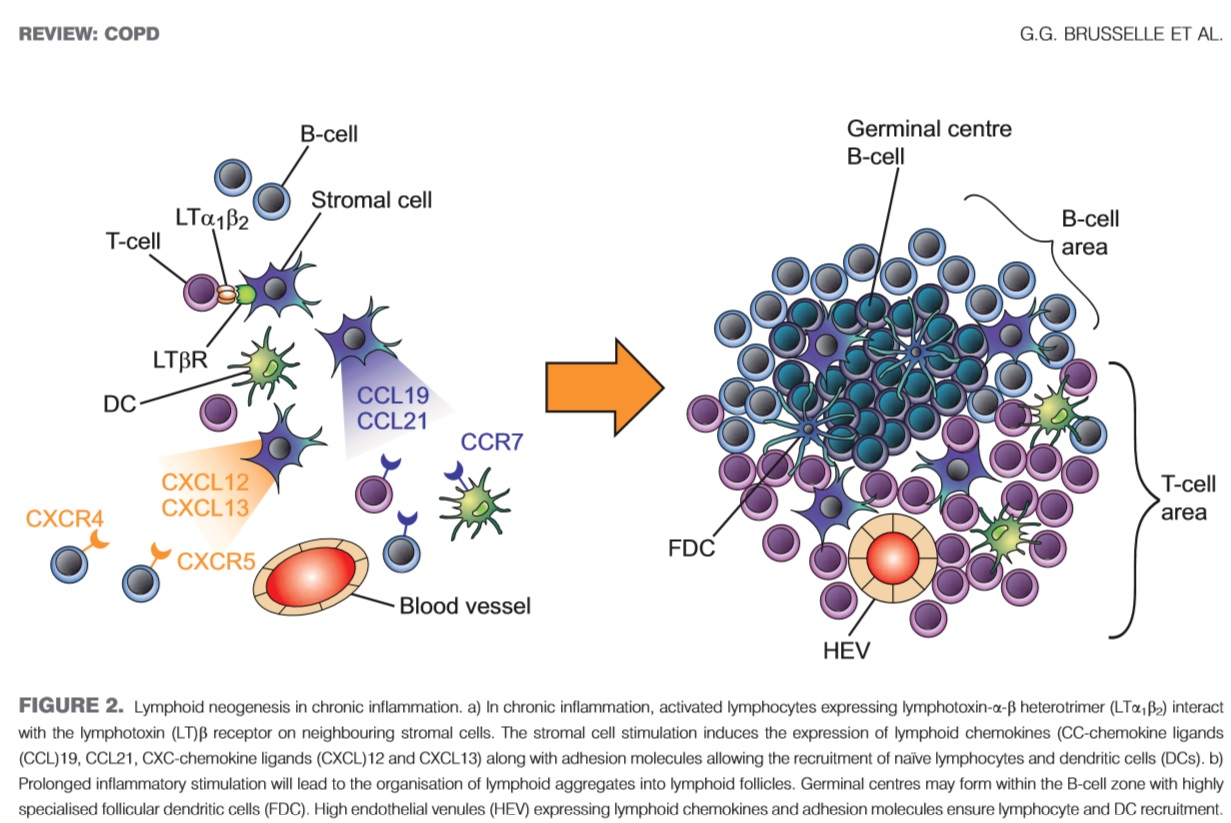
سلول های B CELL
ارتشاح سلول های B CELL های در راههای هوایی کوچک در بیماران COPD دیده میشود. میزان راههای هوایی کوچکی که حاوی سلول های BCELL و فلیکول های لنفوِیید با مراکز ژمینال در بیماری COPD زیاد میشوند. این مراکز لنفوییدی در5% سیگاری ها با ریه سالم دیده میشود. در 25-30% بیماران با COPD شدید این فلیکول های لنفوییدی دیده میشود. میزان ارتشاح سلول های BCELL در بافت ریه راههای هوایی کوچک در بیماری شدی دیده میشود.
فلیکول های لنفوییدی در راههای هوایی کوچک شامل تجمع سلول های BCELL با دندریتک که با CD4+ ( 80 تا 90 % ) و کمی CD8 محاصره شده است.
و لنفسیت های B آی جی ام را اکسپرس می کنند که نشان می دهد این سلول ها فعال هستند و بخش بزرگ این سلول ها CD27+ هستند که لنفسیت های B خاطره ای هستند. سلول های B همچنین الیگوکولونال هستند حضور فاکتور فعال کننده سلول های B که متعلق به TNF هستند (BAFF ) در درون فولیکول های بیماری های خیلی شدید سی او پی دی دیده می شوند که اطراف CD4 و سلول های دندرتیک و سلول های فیبروبلاستیک هستند BAFF مسئول حیات مداوم سلول های B می باشد.
سلول های تی که CD57 را اکسپرس می کند نشانگر فرایند پیری سلول هاست و در فولیکول های انفوئیدی بیماران COPD افزایش پیدا می کند.
ماکروواش های CD68 در راه های کوچک هوایی بیماران COPD افزایش پیدا می کند و نشانگر شدت بیماری می باشد. ماکروفاژ ها ممکن است در التهاب ریه های این بیماران نقش داشته باشد. ماکروفاژ ها به دو قسم M1,M2 تقسیم می شوند و یک قسمت دیگر آنها فلوتایپ بینابینی دارد که به استروئیدها حساس نیستند. تعداد سلول های دندرتیک در بیماران COPD مورد بحث است در بیماران شدید این بیماری ها مقدار این سلول ها کاهش می یابد. در لاواژ بیماران سیگاری سلول های دندرتیک CD1c اکسپرس می کنند و این سلول ها در بیماری COPD استیبل هم در راه های هوایی کوچک افزایش پیدا می کنند. افزایش سلول های CD1c دندرتیک در راه های هوایی کوچک افراد سیگاری بدون COPD هم دیده می شود.
نوتروفیل ها در خلط و لاواژ بیماران COPD استیبل زیاد می شوند و پروتئین 1 آلفا MIP ترشح می کنند. این پروتئین در بیماری شدید COPD نسبت به بیماری متوسط آن افزایش پیدا می کنند و موجب ارتشاح ماکروفاژها می شود. افزایش سلول های نوتروفیل و ماکروفاژهای CD68 در بافت زیر موکوسی دیده می شود و تعداد سلول های تی CD3+ کاهش پیدا می کند. از آنجا که اینترلوکین 8 موجب ترشح مایلوپروکسیداز یا (MTO ) از نوتروفیل ها می شود که خود باعث افزایش سلول های التهابی می شود. افزایش نوتروفیل ها در راه های هوایی کوچک بیماران شدید COPD افزایش پیدا می کند. در عوض پاسخ ایمنی سلول های تی در بیماران شدید COPD مختل می شود.
بر خلاف آسم اکثر مطالعات نشان می دهد که تعداد سلول های مست سل و ائوزینوفیل در دیواره ی سلول های کوچک COPD ی افزایش پیدا نمی کند.
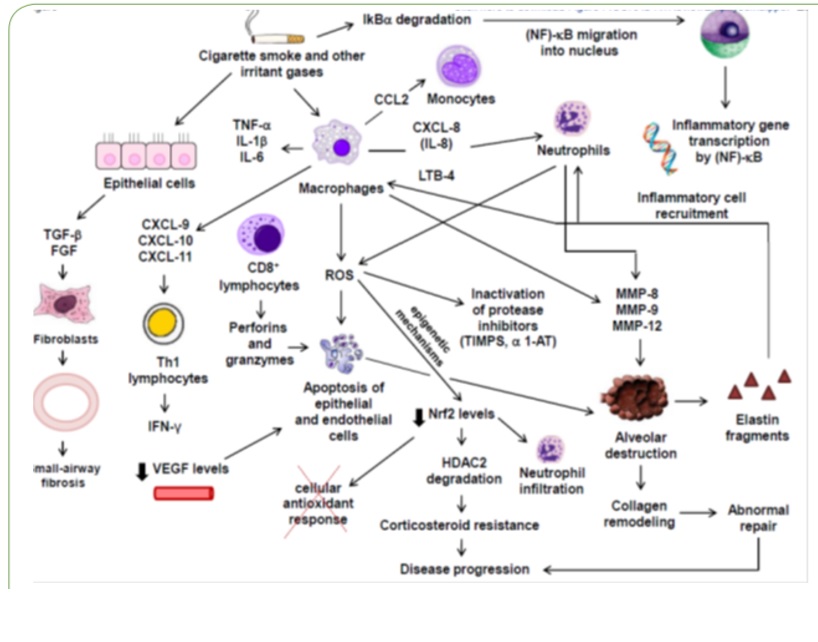
وقتی که فردی سیگار می کشد اول با سلول های پوششی راه های هوایی و سلول های دندرتیک مواجه می شود. این سلول ها هم مواد که موجب ارتشاح سلول های التهابی می شوند. سپس NF-Kb از سلول های پوششی راه های هوایی در بیماران COPD متوسط ترشح می شود که این به نوبه خود موجب افزایش اینترلوکین 1 – 6 – 8 MCP1-TNFα- - ICAM1- می شود
ارتباط بین ترشح راه های هوایی و COPD پیچیده است. خیلی از بیماران برونشیت مزمن افزایش ترشح خلط دارد. وجود برونشیت مزمن خود باعث افزایش مرگ بیماران COPD افزایش ریسک پونومونی و افزایش یا کاهش عملکرد ریه می شود.بسته شدن راه های هوایی کوچک به وسیله این ترشحات در مرگ های زود رس بیماران آنفیزم شدید دیده می شود.
ارتباط مستقیمی بین یکی از موسین ها به نام MUC5AS و میزان سال های سیگار کشیدن وجود دارد. به همین دلیل در ریه هایی که از بیمار درآورده شده شستن راه های هوایی کوچک موجب کاهش مقاومت آن راه ها می شود. مقدار موسین های بی طرف در بیماران COPD منجر به انسداد راه های هوایی کوچک و افزایش پونومونی در COPD می شود.
نقش سلول های آندوتلیوم
در آزمایشات آزمایشگاهی میزان استیل کولین با میزان تمایز TH17 هماهنگی دارد. مولکول های اتصالی مثل ELAM-1 در آندوتلیوم بیماران ملایم تا متوسط COPD زیاد می شود و این ارتباطی دارد با به کار گیری و ارتشاح موضعی نوتروفیل ها در بیوبسی برونشیال بیماران با COPD شدید مقدار زیادی اینترلوکین 22 در اپیتلیال و اینترلوکین 22 و 17 زیر موکوس در بیماران COPD ملایم تا متوسط وجود دارد. این نشان می دهد نقش TH17 و سایتوکان های ترشح شده توسط این سلول ها در ماندگاری نوتروفیل های بافتی در این بیماران. در این مطالعه کمتر از 5 درصد از سلول هایی که اینترلوکین 17 را داشتن تی سل بودند و 90 درصد آندوتلیال (CD31+ ) بودند.همچنین 10 درصد از سلول هایی که اینترلوکین 22 داشتن تی سل بودند و 80 درصد آندوتلیال بودند. بنابراین نقش عمده در تولید اینترلوکین 17 را آندوتلیال ها دارند که باعث می شوند این سایتوکاین ها در سلول های رونشیال در بیماران COPD افزایش پیدا کند. کموکاین ها مثل CCL5,CXCL7 در واقع موجب ارتشاح نوتروفیا ها می شود و در بیماری شدید COPD دیده می شود و باز نقش مهم آندوتلیال در شدت بیماری COPD را نمایان می کند.
در مطالعاتی میزان آپوپتوز در سلول های آندوتلیال و اپی تلیال در محیط ریه افزایش می یابد در نتیجه این فرایند آمفیزم ریوی ایجاد می شود. همچنین VEGF در پوشش برونشیول ها در بیماران COPD کاهش پیدا می کند. نقص عملکرد سلول های آندوتلیال شریان های ریوی کوچک همراه است با افزایش رشد و مواد که باعث انقباض عروق می شود در نتیجه موجب ضخامت اینتیما و ارتشاح CD8 می شود. افزایش فشار شریان ریوی در بیماران COPD شایع می باشد و وجود آن و شدت آن و شدت بیماری و پیش آگهی بیماری ارتباط دارد.همچنین تغییر آناتومیک سلول های عروقی با شدت کاهش اکسیژن شریانی ارتباط دارد.
درمان های COPD :
برخلاف آسم که درمان کورتنی خیلی موثر است ولی در COPD در کاهش التهاب راه های هوایی و کاهش عملکرد ریه نقش زیادی ندارد اما استفاده از کورتن های استنشاقی مثل بودزناید و فلوتیکازون همراه با لابا همراه است با افزایش احتمال عفونت های جدی اما در نهایت موجب افزایش مرگ و میر نمی شود. در یک مطالعه اخیر نشان داد که کورتن های استنشاقی و کاهش CD4,CD8 موثر هستند و باعث کاهش نوتروفیل ها و لنفوسیت های ریه می شوند اما میزان ماکروفاژهای ریه را افزایش می دهند. مطالعه دیگری نشان داد که میزان CXCL8 در لاواژ بیماران کاهش پیدا می کند. سه ماه استفاده از سروتاید باعث کاهش CD8,CD68 در بیوبسی برونشیال در COPD های متوسط تا شدید شد. درمان بین شش تا سی ماه با یا بدون گشاد کننده لوله های ریه موجب کاهش CD3,CD4 مست سل ها در COPD های متوسط تا شدید شد.همچنین کورتن های استنشاقی باعث کاهش سلول های دندرتیک ماکروفاژها و نوتروفیل ها در افراد سیگاری می شود.
ما می دانیم که در بیماران COPD پروتئین های افزایش دهنده التهاب زیاد می شود و سایتوکاین های کاهش دهنده التهاب مثل اینترلوکین 10 کاهش پیدا می کند. هم گونه های واکنشی اکسیژن (ROS ) و هم گونه های واکنشی نیتروژن (RNS ) در ایجاد بیماری COPD نقش دارد. همچنین سلول های التهابی در بیماران COPD باعث افزایش اکسیدان ها می شود. اکسیدان ها باعث رشد و افزایش سلول ها می شوند. که در نهایت موجب نکروز و آپوپتوزیز سلول ها می شوند. این فرایند ها می توانند در تماس با آلودگی های هوا و سیگار ایجاد شوند.تنها قسمتی از سیگاری ها مبتلا به COPD می شوند بنابراین می توان فرض گرفت که دفاع آنتی اکسیدانی بر اکسیدان ها در این بیماران غلبه می کند.
می دانیم که سایتوکاین هایی مثل TNF-a, IFN-ɣ - IL1β – IL6- IL17- IL18 – IL32 – TSLP- فاکتورهای رشد مانند TGF-β زیاد می شود. درمان های ضد سایتوکاین ها فعلا در مطالعه هستند و برخی از آنها به علت عوارض زیاد مثل عفونت های ریه قطع شده اند.
ایمونولوژی حملات COPD :
در حملات COPD میزان نوتروفیل های خلط افزایش پیدا می کند که خود باعث نارسایی شدید تنفسی با یا بدون عفونت می شود. در عوامل ویروسی ایجاد کننده حملات ویروسی افزایش ائوزینوفیل ها در خلط دیده می شود. افزایش CD8 در حملات COPD با کاهش اینترفروم گاما و اینترلوکین 4 که نماینده CD4 هستند دیده می شود. بنابراین یک انتقال به سمت Tc2 در حملات COPD دیده می شود که باعث افزایش ائوزینوفیل ها می شود به خصوص در حملات این بیماری در اثر ویروس. بیماران با حملات ملایم تا متوسط افزایش ائوزینوفیل ها را در پوشش راه های هوایی نشان می دهد همچنین افزایش CCL5 نوتروفیل ها لنفوسیت ها تی ان اف آلفا دیده می شود. در حملات شدید COPD همراه است با افزایش نوتروفیل ها CXCL8 – CXCL5 – CXCR1,2 . پس نوتروفیل ها در بیماری های شدید COPD ملاحظه می شود و در حملات ملایم تر ائوزینوفیل ها زیاد می شود. از این جهت با آسم متفاوت است که حضور ائوزینوفیل ها موقتی است و در عروق مویرگی دیده می شود.
یک مطالعه نشان داد که رینو وایروس ها می توانند موجب حملات COPD شوند و میزان لازم این ویروس ها برای حملات در این بیماران کمتر از بیماران نرمال است. نوتروفیل های خلط بیماران COPD بعد از غفونت افزایش پیدا می کند. میزان CD3 متناسب است با میزان مقدار ویروس ها در حملات همچنین اینترلوکین 27 و 10 هم افزایش پیدا می کند . عفونت های باکتریایی مثل هموفیلوس آنفولانزا در 60 درصد از بیماران COPD بعد از ویروس رینو وایروس دیده می شود. مقدار الاستاز نوتروفیلی خلط افزایش پیدا می کند.