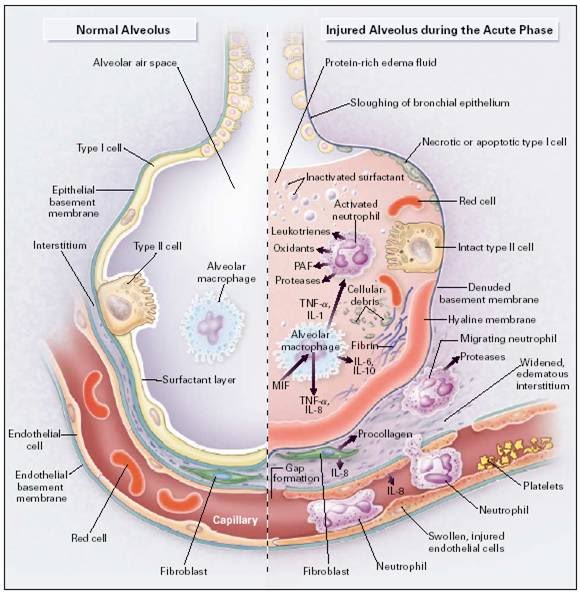
ایمنوپاتولوژی ARDS :
این بیماری باعث مرگ و میر زیادی به علت تجمع مایع پروتئینی در آلوئول ها می شود. شایع ترین علت آن ویروس ها و باکتری ها هستند و به ندرت قارچ. در شرایط دیگری مانند سپسیس سوختگی تروموهای زیاد و پانکراتیک می توانند ایجاد شوند. ایجاد این بیماری هم ایمنی ذاتی هم ایمنی انطباقی دخالت دارد.
ایمنی ذاتی:
سلول های اپیتلیال آلوئول در واقع یک محافظ آلوئول هستند ماکروفاژها در درون این فضا قرار می گیرند. و فضای بینابینی هم اولین جایی است که صدمه یا میکروب را درک می کنند. سلول های دندرتیک یا TC بین ایمنی ذاتی و انطباقی قرار دارند و محلشان در لایه های اپیتلیال در دیواره های آلوئولی و پارانشیم ریه می باشد. ضایعه های بلندی دارند که می توانند آنتی ژن های بلع شده را تست کنند. و سپس پاسخ انطباقی را ایجاد کنند در مجموع سلول های اپیتلیال ، ماکروفاژها و دندرتیک سل ها مانند یک واحد سلولی حس گر عمل می کنند و می توانند سیگنال های خطرناک را با واسطه PRR و پاسخ اولیه ذاتی مهار کنند. اگر خطر عفونی باشد به آن PAMP می گویند و اگر غیر عفونی باشد DAMP یا آلارمین می باشد. توانایی حسی این سیگنال ها وابسته است به رسپتورهای تول – لایک (TLR) . سلول های پوششی آلوئول اینترفرون گاما CCL2 ترشح می کنند. ماکروفاژها اینترفرون بتا IL6 – TNF- IL12 ترشح می کنند و دندرتیک ها IL12 – IL23 – IL1β ترشح می کنند. لنفوسیت های بافتی هم شامل تی سل های سایتوتوکسیک TH1 – TH17 – NKCELLS – NKTCELLS -ɣΔtcell و سلول های لنفوئیدی ذاتی از نوع یک تا سه می باشد. گروه یک این سلول های لنفوئیدی سایتوکاین های TH1 شامل اینترفرون گاما و تی ان اف ترشح می کنند. گروه دوم ILC سایتوکاین های TH2 را ترشح می کنند که شامل اینترلوکین 5 – 13 و 33 می باشند. در حالیکه گروه سوم این ILC ها سایتوکاین های TH17 را ترشح می کنند مانند IL17A – IL22 . سلول های تی ساتوتکسیک تحریک شده و فعالیتشان افزایش پیدا می کند. NKCELL ها اینترفرون گاما و 22 ترشح می کنند باعث حمله به ویروس ها و باکتری هایی که با ویروس ها عفونتی شده اند می شود. در نهایت باعث تحریک دندرتیک سلول ها می شوند.گاما دلتا سل ها منشا تولید اینترفرون گاما – تی ان اف آلفا و اینترلوکین IL17 هستند این ها سایتوکاین های موج دوم هستند که باعث تجمع نوتروفیل ها و منوسیدها به داخل ریه می شوند.
در یک فرد سالم ماکروفاژها نقش هموستازیز اجرا می کنند بدون اینکه موجب شوند التهاب ایجاد شود. این ها شامل تولید GM-CSF برای تولید سورفاکتانت که بدون آن ذرات در آلوئول تجمع پیدا می کنند و موجب مایع پروتئینی در آلوئول می شوند. در شرایط ایده آل ماکروفاژها سیگنال های منفی شامل IL10 – TGFβ و CD200 از سلول های پوششی آلوئولی مرتبا دریافت می کنند. TGFβ و اینترلوکین 10 باعث تحریک رسپتورهای CD200 می شوند. TGFβ همراه با سلول های اپیتلیال ماکروفاژها را مهار می کنند. به خصوص در ترشح نوع 1 اینترفرون نسبت به آنفولانزا
در طی بیماری ARDS ماکروفاژها بین فنوتایپ ضد التهابی M1 و ضد التهابی M2 قرار دارند. منوساید ها عمدتا ماکروفاژهای التهابی ایجاد می کنند. ولی ماکروفاژها در حضور LPS خصوصیت ضد التهابی از خود نشان می دهند. در این مطالعه که روی موش ها صورت گرفت کاهش منوسیدها باعث کاهش نوتروفیل ها و IL17 – MCP1 – HMGB1 شد. از بین بردن ماکروفاژها باعث افزایش این مدیتورها شد. بنابراین این تصور وجود دارد که ورود منوسیدها با شدت نارسایی تنفسی که نیاز به اکسیژن زیادی دارد تناسب دارد. مطالعات بیشتر نشان داد که روی M1 – ACAM1 – CD40 و روی M2 رسپتور ترنسفرین و مانتوز وجود دارد. M1 توسط سایتوکاین های TH1 تعریف می شود مانند اینترفرون گاما و LPS . و بعد خود آن اینترلوکین 1 بتا ، اینترفرون 12 ، تی ان اف آلفا و NOS ترشح می کند. در مقابل M2 توسط سایتوکاین های TH2 مانند اینترلوکین 4 و 13 تحریک می شوند و اینترلوکین 10 را ترشح می کنند. مثلا تزریق APS همراه با M2 موجب کاهش ارتشاح نوتروفیلیک استرس های اکسیداتیو، تی ان اف آلفا ، IL6 و IL1β شده و برعکس موجب افزایش TREGULATORY (TREG) که در نهایت اینترلوکین 10 را ترشح می کنند. اگر PD1 یا PDL1 را بلاک کنیم سایتوکاین های M2 در لاواژ بیماران برعکس می شوند. درمان با متیل پردنیزولون باعث می شوند که M2 غالب شود.
نوتروفیل ها در خون جریان دارند و در تماس با CXCL8 وارد بافت ریوی می شوند. نوتروفیل ها MMP9 ترشح می کنند که باعث از بین رفتن کلاژن ها در ECM می شود. نوتروفیل ها سپس شروع به فاگوسیت و رها کردن نوتروفیل پلاستاز مایلوپروکسیداز ROS – NET که هدف همه آنها از بین بردن ویروس ها باکتری ها و قارچ ها می باشند. به این فرایند نتوزیز می گویند. که در آن مرگ نوتروفیل ها اتفاق می افتد توسط ایجاد NETS شاما پیستون فیبرهای کروماتین می باشد و هدفش مهار پاتوژن هاست. اما این شبکه NETS اگر مهار ناپذیر باشد موجب از بین رفتن بافت ریه می شوند. ویروس RSV می تواند ایجاد NET بکند و به خوبی ویروس را مهار بکند اما وقتی این عمل در داخل بدن صورت می گیرد موجب تشکیل پلاگ های سفت NET می شود که در نهایت موجب بسته شدن راه های هوایی بدون از بین بردن ویروس ها می شود.
برخی باکتری ها مانند استافیلوکوک آئوروس، استرپ پونومونی و هموفیلوس آنفولانزا می توانند شبکه NET را خنثی کنند. به علاوه افزایش عفونت باعث می شود که کلاسترهای NET بیشتری بر علیه باکتری ها و آنفولانزا ها تشکیل بشود و موجب تشدید التهاب – افزایش باکتری ها و در نهایت نقص تنفسی بشود. این NET ها که در داخل آلوئول ها قرار می گیرند نقش نتوزیز و ARDS را توضیح می دهند.
در شرایطی که صدمه ریوی بافتی ناشی از تزریق خون پلاکت های فعال شده باعث تشکیل NET می شوند که در نهایت باعث تشکیل لخته در ریه می شود. NET ها در عروق بسیار ریز دیده می شوند در حالی که در مورد ARDS ، NET ها در آلوئول ها تشکیل می شوند.
یک مطالعه بر روی تایروفیبان نشان داد که میزان NET ها را کم می کند. موجب کاهش مایع ریوی می شود و کاهش انتقال پذیری مایعات در مطالعه انسانی میزان پایین دوز آسپرین باعث کاهش نوتروفیل ها و پروتوآزهای آنها مثل MMP8 و MMP9 در لاواژ شد.

سیستم انطباقی:
دندرتیک سل ها بعد از حس کردن پاتوژن ها بر روی CD4 تی سل ها اثر می کنند و CD4 در پاسخ تبدیل به TH1 یا TH2 می شود. TH1 تحت تاثیر فاکتورهای ترجمه ای مثل T-BET و STAT4 و IL12 قرار می گیرند.و باعث مرگ سلول های عفونی از طریق اینترفرون گاما می شوند.TH2 تحت تاثیر فاکتورهای ترجمه ای گاتا 3 و استت6 و اینترلوکین 4 قرار می گیرند و باعث تحریک سلول های بی سل و تولید آنتی بادی می شوند. CD8 تی سل در پاسخ با دندرتیک سل ها به سلول های سایتوتوکسیک یا حافظه ای تبدیل می شوند. در یک مطالعه نشان داد که ترشح LPS به یک موش باعث باعث ترشح CD8 ها ظرف 12 ساعت انجامید. بیمارانی که میزان ویروس پایینی دارند معمولا CD8 آنها از CD4 آنها بیشتر است. بر خلاف بیمارانی که اصلا عفونت ندارند. CD8 ها آنتی ژن را با کوپلکس MHC تشخیص می دهند و باعث از بین رفتن سلول های هدف با تشکیل پرفورین و گرنزین می شوند. پرفورین یک سوراح ایجاد می کند و گرنزین هم وارد سلول هدف می شود. به علاوه سلول های سایتوتوکسیک اینترفرون گاما تی ان اف آلفا و IL2 ترشح می کنند که باعث می شود تعداد خود سلول ها و طول عمر آنها افزایش پیدا کند همراه با TREG ها سلول های ساپوتوکسیک اینترلوکین 10 را هم ترشح می کنند که بدون آن التهاب می تواند خارج از کنترل قرار بگیرد. سلول های حافظه ای اینترلوکین 22 – 17 و اینترفرون گاما را ترشح می کند.
TH17 تولید IL17A – IL17F –IL21 –IL22 می کنند. شاخصه ی IL17 تحریک CXCL9 – 10 برای ارتشاح نوتروفیل ها و افزایش آنها می باشد. در واقع در نبود IL17A و حتی در حضور LPS میزان ارتشاح نوتروفیل ها به مراتب کمتر می شود. کاهش TH17 همچنین باعث کاهش لیک پذیری سلول های اپیتلیال می شود. مطالعه دیگر نشان داد که کاهش IL17 , 22 موجب کاهش ارتشاح نوتروفیلی و ماکروفاژی می شود و باعث افزایش سلول های TREG می شود. به خاطر همین هست که بیماران ARDS افزایش IL17A در لاواژ انها دیده می شود.
نقش TREG ها در ARDS :
TREG سلول هایی هستند که با CD25 اکسپرس می شوند و فاکتورهای ترجمه ای هم هست FOXP3 که در واقع با ترشح IL10 , TGFβ که باعث هموستازیز می شوند. در صورتی که این سلول ها وجود نداشته باشند ما شاهد افزایش التهاب و TH17 خواهیم بود. بیماران ARDS ی که بهبود پیدا کرده اند اینترلوکین 10 بیشتری ترشح کرده اند. خصوصیت ضد التهابی TREG ها از بین بردن خصوصیت درک آنتی ژنی منوسیدها و دندرتیک ها می باشد. تعادلی بین TH17 و TREG در مطالعات مرگ و میری بررسی شده است. افزایش TH17 نسبت به TREG ها در 24 ساعت اولیه ARDS همراه است با بدخیم بودن بیماری. TREG ها همچنین در ساختمان سازی هم نقش دارند و سلول های اپیتلیالی آلوئولی که تخریب شده اند را تعمیر می کنند. این روند توسط کراتینوساید جی اف که توسط TREG ها که در فاز بهبودی ترشح می شوند ایفا می شود. این موضوع باعث شده است که برخی داروها که باعث افزایش TREG ها می شوند و موجب تسریع فرایند ترمیم می شوند مورد بررسی قرار بگیرند. مثلا آنتیبیتیک هایی مثل ماکرولیت ها – اریترومایسین باعث افزایش تعداد TREG ها می شود. و کاهش صدمه ی بافتی. این باعث می شود که تناسب TREG ها به TH17 تصحیح شود. PD1یک پروتئینی است که در ساپرشن پاسخ مزمن تی سلی نقش دارند و می تواند با کمک TREG سل ها در مهار ARDS نقش داشته باشد. PDL1 بعدا مشخص شد که در پاسخ TREG ها نقش مهمی را دارد.
CTLA4 یک پروتئین دیگر است که روی TREG ها اثر می کند و باعث یک تولید مثل تی سل ها به شکل منفی می شود. CTLA4 روی CD4 , CD8 در بیماران ALI بالاتر می رود.و در زمان مرگ میزان اکسپرسیون این پروتئین بیشتر از زمان ارتشاح آن است. یکی از مسائل دیگر مسئله استفاده از سلول های بنیادی مزانشیال می باشد که باعث تسریع التهاب می شود.در حال حاضر یک مطالعه به نام START در فاز دوم قرار دارد. مسئله بعدی گاما دلتا تی سل ها در پاسخ به عفونت در ریه افزایش پیدا می کنند و تولید اینترفرون گاما و IL17 را فزایش می دهند.
یکی از تکنیک های جدید که امروز در ARDS به وجود آمده است سایتومتری توسط TIME – OF – FLIGHT (CYTOF) در این تکنیک ماکروفاژهای ARDS از ماکروفاژهای NONARDS توسط شناسایی CD169 و PD-L1 تشخیص داده می شوند.ماکروفاژهایی که هردوی آنها را زیاد اکسپرس می کنند AM1 هستند آنهایی که CD169high زیاد و PDL1LOW کم AM2 نامیده می شوند و آنهایی که CD169LOW دارند AM3 نامیده می شوند. اولی ماکروفاژ سالم است دومی ماکروفاژ غیر ARDS ی است و سومی ماکروفاژ ARDS می باشد. بنابراین وجود اکسپرسیون PDL1 می تواند در آینده بیماری ARDS نقش داشته باشد.
در کرونا آنتی بادی هایی وجود دارد که حتی قبل از تماس با ویروس به وجد آمره است این آنتی بادی ها از نوع IGM هستند این آنتی بادی ها به شکل سریع بعد از سن 40 سال و به خصوص در مردان و آنهای که گروه خونی A دارند کاهش پیدا می کند. البته تا کنون مطالعات مختلف نتایج مختلفی را داده اند. مسئله دیگر اینترفرون ها هستند . ایترفرون ها پاسخ بسیار مهمی در مقابل ویروس ها ایجاد می کنند و در مطالعات پیشین اینترفرون 1 و 3 نقش مهمی در مهار سارس – COV1 داشتند. اینترفرون تایپ 1 و 3 وقتی که سلول با ویروس عفونت پیدا می کند ایجاد می شود.
تایپ 1 اینترفرون شامل اینترفرون آلفا / بتا می باشد.در بیماری قبلی کووید 1 یک تاخیر در تولید اینترفرون نوع یک باعث شد که التهاب ماکروفاژی و منوسیتی ایجاد شود. در کووید 19 هم احتمالا چنین ناهنجاری هایی در تولید اینترفرون نوع یک و سه وجود دارند. و این بیماران اینترفرون یک و سه کمتری ترشح می کنند. کمبود این اینترفرون ها باعث افزایش میزان ویروس و در نتیجه افزایش پاسخ ایمنی همراه با TNFα و IL6 می شوند.یک مطالعه نشان داد که بعضی از خود ایمنی ها بر علیه اینترفرون نوع 1 می تواند 10 درصد کرونایی های شدید را توجیه کند. نوع 3 اینترفرون یا همان اینترفرونLAMBDA است و خصوصیت های مشابهی با نوع اول اینترفرون و اینترلوکین 10 دارد. این نوع اینترفرون بیشتر در مخاط ریه ها و گوارش دیده می شود. تایپ یک رسپتورها در همه سلول ها وجود دارد ولی تایپ سه رسپتورها فقط در سلول های اپیتلیال نوتروفیل ماکروفاژ بی سل و دندرتیک ها وجود دارد.این پیشنهاد می کند که تایپ 3 اینترفرون ها بیشتر پاسخ مخاطی می دهند. تایپ 3 اینترفرون ها زودتر ترشح می شوند. ممکن است بتوانند جلوی رشد ویروس را بگیرند. آنها پتانسیل ایجاد التهاب مثل اینترفرون تایپ 1 را ندارند و عملا خصوصیت ضد التهابی دارند. مطالعات اخیر در حیوانات نشان داده است که تایپ یک و سه اینترفرون در مهار لوکال یا تایپ سه در واقعدر مهار سیستمیک نقش دارند. اما در عین حال پیشنهاد شده است که تایپ یک اینترفرون ممکن است باعث ایجاد یک التهاب شدید در ریه بشود. مطالعه نشان داده که از آنجایی که ویروس کرونا یک پاسخ خوب اینترفرونی یک و سه ایجاد نمی کنند ولی می توانند تولید کموکاین های زیادی بکنند در نتیجه ممکن است ایجاد ارتشاح التهابی بکنند. مثلا در مطالعه ای در موش ها که کرونا گرفته بودند نشان داده شد که کموکاین های جذب کننده منوساید ها و نوتروفیل ها که شامل CCL2 – CCL8 – CXCL8 – CXCL12 بکنند. همچنین سن می تواند عامل مهمی در این اختلال از سمت دفاع به سمت التهاب نقش داشته باشد. مثلا کسانی که 65 سال بالاتر داشتند و کرونا گرفتند 80 درصد بیمارستانی می شوند و احتمال مرگشان 23 برابر می شوند. البته بیماری های دیگر با سن را هم شامل دیابت بیماری های قلبی و چاقی را هم باید در نظر گرفت به هر حال مانند هر ارگان دیگری سیستم ایمنی با سن تغییر پیدا می کند. دو تغییر بزرگ ایمنوسن اسنس و اینفلامیجینگ هست. ایمنوسن اسنس یک کاهش عملکردی در سیستم ایمنی است که قرار است با پاتوژن ها بجنگد. این باعث می شود که بیمار مستعد عفونت ها بشود و مستعد بیماری های مزمن از جمله خود ایمنی و سرطان بشود. اینفلامیجینگ یک افزایش التهاب سیستمیک است که باعث می شود که این تغییرات در سیستم ایمنی ایجاد شود و سایتوکاین های التهابی ایجاد بشود.
مسئله دیگر اینترلوکین یک می باشد. که اساسا توسط فاگوسیت های منونوکلئار ترشح می شود و می تواند موجب ترشح IL6 و TNFα بشود. گرچه میزان ترشح جزئی از IL1 می تواند دفاعی باشد اما میزان زیاد آن می تواند مرگ آسا باشد.همچنین امروزه مشخص شده که IL1 با IL33 , 18 در ارتباط هست.
آنتی بادی ها:
اکثر مواقع IGM بین 8 تا 12 روز از عفونت در سرم پیدا می شود و در عرض 12 هفته هم از بین می رود. IGG دو هفته دیرتر تشکیل می شود و طولانی تر هم باقی می ماندو پاسخ IGG وابسته است مقدار ورودی ویروس ها و شدت بیماری. مطالعه نشان داد آنهایی که بیماری بدون علامت خفیف داشتند تولید IGG کمتری کرده اند. البته این مسئله در قضیه ایمنی مورد بحث قرار دارد ولی برخی معتقدند کسانی که بیماری ملایم دارند ممکن است نتوانند ایمنی طولانی از خود به جا بگذارند. در زمانی که پلاسما های افراد بهبود یافته استفاده شد نشان داده شد که پلاسماهای حاوی آنتی بادی خصوصیت متوسط بر ضد ویروس دارند. و این پاسخ بین روز دهم تا پانزدهم بیماری اتفاق می افتد.بنابراین از آنجایی که ممکن است آنتی بادی های خنثی کننده تا 2 هفته پس از ترخیص از بیمارستان تولید نشود ممکن است این بیماران از این آنتی بادی های خنثی کننده سود ببرند. مطالعات اخیر نشان داده است که بیمارانی که نقص اولیه اومرال دارند و آنتی بادی تشکیل نمی دهند. کرونای شدیدی هم پیدا نمی کنند. این مسئله نشان می دهد که ایمنی ذاتی در از بین بردن ویروس نقش مهمی را بازی می کند.
سلول های بی:
IL6 و TNFα با تنظیمات سلول های بی ارتباط دارند تغییرات در زیر مجموعه های سلول های بی در کرونا دیده شده است. که یکی از آنها افزایش پلاسمابلاست ها و کاهش سلول های حافظه ای بی می باشد. و برخی مطالعات پلاسمابلاست ها 30 درصد از سلول های بی را تشکیل می دهند. عدم وجود ارتباط بین پلاسمابلاست ها و آنتی بادی های خنثی کننده وجود دارد. تی سل ها نقش مهمی در ایمنی انطباقی در مقابل کرونا دارند. CD8 ها که می توانند سلول های عفونی را هم از بین ببرند در حالی که CD4 ها می توانند CD8 ها را فعال کنند.
اکثرا کاهش لنفوسیت ها شامل سلول های تی و گاها سلول های بی و NCELL هاست. کاهش لنفوسیت های سی و NKCELL ها خیلی بیشتر از سلول های بی است. بعضی از بیماران شدید حتی افزایش سلول های بی را نشان می دهد. بنابراین سلول های تی در پروسه عفونت کرونا بیشتر صدمه می بینند. این احتمالا به این خاطر است که سلول های تی ایمنی سلولی بر غلیه ویروس را هدایت می کنند در حالی که سلول های بی تولید کننده آنتی بادی ها برای خنثی کردن ویروس ها هستند. بیشترین آنالیز ها مربوط به کاهش سلول های CD4,CD8 می باشد. در بیمارانی که دچار ARDS می شوند تقسیم CD8 به CD4 کاهش پیدا می کند. یافته های زیادی نشان میدهد که کاهش لنفوسیت ها با شدت بیماری و مرگ و میر ناشی از بیماری ارتباط دارد. در بیماری های ملایم تعداد لنفوسیت ها نرمال هستند و یا در طول درمان به نرمال می رسند. کاهش لنفوسیت ها در بیماران شدید در روز 4 تا 6 بعد از بیماری به حداقل می رسد. کاهش کمتر از 800 لنفوسیت به ازای یک میکرولیتر می تواند نشانه ای از این باشد که بیماری نیاز به مراقبت بیشتری دارد تا وارد ICU نشود. وقتی که لنفسیت ها در مورد روز پانزدهم بعد از درمان به حد نرمال میرسند بیماران بعد از 25 روز شفا پیدا می کنند. از آنجایی که رسپتورهای این ویروس در روی سلول های تی کم است در نتیجه احتمال کاهش لنفوسیت ها به علت حملات مستقیم ویروس کم می باشد. علت اول کاهش لنفوسیت ها ممکن است ناشی از از بین رفتن لنفوسیت ها در طی این بیماری باشد. نامنظم شدن سایتوکاین ها ممکن است به مرگ لنفوسیت ها کمک بکند. تا کنون مشخص بوده است که TNFα و اینترلوکین 6 عامل مهمی در نابودی سلول ها می باشد. به خصوص TNFα با اتصالش به رسپتور خودش روی سلول های تی می تواند منجر به نابودی سلول های تی شود. مرحله دوم تزریق استروئید ها و داروهای ضد ایمنی به بیماران شدید کووید می تواند موجب برگشت لنفوسیت ها شود در نتیجه موجب تقویت از بین رفتن سلول های ویروسی شود از طرف دیگر لنفوپنی ها ممکن است ناشی از از بین رفتن ارگان های لنفاتیک مثل تیموس و فعال باشد.حتی اسیدوز لاکتیک هم می تواند به لنفوپنی منجر شود بیماری کرونا وقتی همراه با HIV می شود که یک کشنده سلول CD4 می باشد می تواند موجب افزایش طول مدت کرونا به مدت دو ماه بشود.
مسئله بعدی این هست که لنفوسیت های باقی مانده هم از نظر عملکرد مختل می باشد. سلول های زنده تی سل بعد از عفونت میزان زیادی PD1 و TIM3 که هردو نشان دهنده از پا در آمدگی و افزایش بیماری است نشان می دهد. همچنین این سلول ها CTA4 و TIGIT را اکسپرس می کند. خستگی نچرال NKCELL ها و CTL ها در تولید اینترفرون گاما – اینترلوکین 2 – گرنزیم بی – تی ان اف آلفا و CD107A دیده می شود. در مرحله بهبودی این عملکردها به جای مناسب بر می گردد. در به وجود آوردن این عملکرد ضعیف اینترلوکین 10 مهم تلقی شده است. در واقع با مهار اینترلوکین 10 می توان جلوی خستگی سلول های تی را گرفت. از آنجایی که افزایش اکسپرسیون PD1 و CTAL4 و TIGIT و NKG2A و TIM3 همراه هستند با خستگی سلول های لنفوسیت مهار این ها ممکن است باعث افزایش فعالیت لنفوسیت ها در مراحل اولیه بشود.
در اکثر موارد وایت بی سی ها و نوتروفیل ها در بیماری کووید بالا می رود و در بیماران شدید میزان وایت بی سی ها و نوتروفیل ها بالاتر هم می رود ممکن است این افزایش در طول سه روز اول اتفاق بیفتد و به زودی به حد نرمال برگردد. نسبت نوتروفیل ها به لنفوسیت ها یک معیاری است برای شدت بیماری. بیمارانی که این نسبت بالایی دارند میزان اینترلوکین 2 – 6 و 10 آنها بالاتر است نشان دهنده این است که بیماری کووید آنها شدیدتر می تواند باشد.
پروتئین S روی ویروس شامل S1,S2 و RBD بسیار ایمونوژیک هستند و می توانند مورد حمله توسط آنتی بادی ها قرار بگیرند. آنتی بادی اختصاصی به نوکلئوکسپید که در بیماری کووید دیده می شود خصوصیت نوترولایزینگ ندارد. CD4 موجب تولید آنتی بادی ضد ویروس می شود که در فعالیت بیش از حد پلاسما سل ها در لاواژ بیماران کوویدی دیده می شوند. در بیماران پیر دیده شد که آنتی بادی های خنثی کننده ضد S پروتئین بالا بود و فعالیت بیشتری نسبت به جوان ها داشت کاهش سلول های لنفوسیتی در بیماران مسن با میزان تیتر آنتی بادی های نوترولایزینگ هماهنگی دارد. بنابراین به نظر می رسد که افزایش واکنش ایمنی اومورال پاسخی باشد به نقص ایمنی سلولار .
IGM – IGG – IGA آنتی بادی هایی هستند که به تدریج بالا می روند در حالی که IGG1, IGG3 ، IGG های اصلی IGG هستند. ظهور IGM و IGG ها در روز هفتم نا نوزدهم بعد از شروع بیماری دیده می شوند. تقریبا صد در صد بیماران IGG مثبت هستند و 94 درصد IGMمثبت هستند. IGM شروع می کند به کاهش دو تا سه هفته بعد از شروع علائم. بیماران شدید میزان کمپلمانت C3 زیادی دارند. در نتیجه مهار کمپلمان C3 ممکن است باعث کاهش التهاب در ریه بشود. در حال حاضر داروهای ضد کمپلمان C3 و ضد کمپلمان C5 در حال مطالعه می باشند. در بیماران شدید کرونایی افزایش سایتوکاین ها به خصوص اینترلوکین 1 بتا ، IL2 ,IL4 , IL6 , IL7 , IL8 , IL9, IL10 , IL17, TNFα و اینترفرون گاما و IP10, MIP1α, MCP1, MCP3, G-CSF, GM-CSF دیده می شود. تقریبا دیده شده است که افزایش IL6 در بیماران شدید 10 برابر بیماران دیگر است. افزایش IL6 , IL10 اینترفرون گاما و تی ان اف آلفا می توانند دلایل کاهش سلول های تی باشند. میزان سایتوکاین ها در بیماران شدید به حد ماکزیمم در روز سوم تا ششم بیماری می رسد زمانی که توام است با کاهش شدید لنفوسیت های تی و افزایش اینترلوکین شش و اینترلوکین 10. همچنان که بیماری بهبود می یابد و این سایتوکاین ها نرمال می شوند و این لنفوسیت ها هم به حد طبیعی می رسد.اینترلوکین شش باعث افزایش VEGF می شود که و کاهش ایکدپرین می شود در نتیجه باعث نشت مایعات عروقی به داخل آلوئول ها و ARDS می شود. همچنین بیماری کرونا باعث می شود که روی سیستم نوروایندو کرین اثر می گذارد و باعث ترشح کورتن ها شده که خود باعث ضربه به سیستم ایمنی می شود.
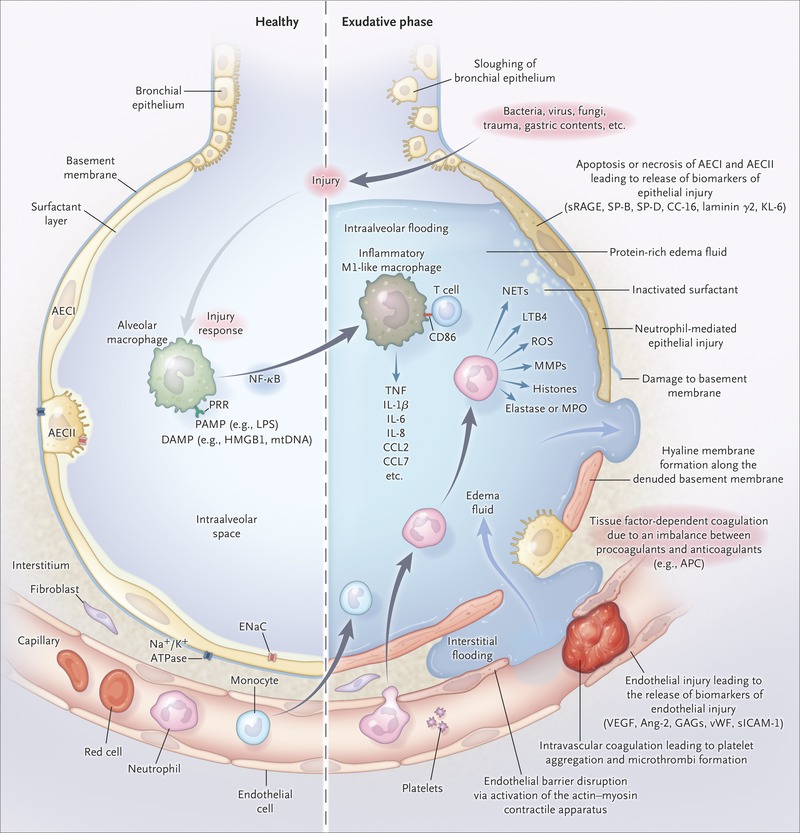
مطالعات قبلی نشان داد که سلول های تی ماکروفاژها و منوسیدها حامل طوفان سایتوکاینی هستند. اما مطالعات جدیدتر نشان می دهد که لنفوسیت های پرفعال ممکن است در مراحل اولیه بیماری در طوفان سایتوکاینی نقش داشته باشند. CD8 به طور کلی اینترفرون گاما ترشح می کنند CD4 به طور شاخص سایتوکاین های TH1 شامل IL2 اینترفرون گاما و TNFα ترشح می کنند و همچنین سایتوکاین های TH2 شامل اینترلوکین 4 و 5 و 9 و 10 و 13 ترشح می کنند اما اینترلوکین 6 از سلول های تی سل نشات نمی گیرد. ماکروفاژها و نوتروفیل ها هم نقش بزرگی در طوفان سایتوکاینی دارند. اکثر سلول های ایمنی می توانند اینترلوکین 10 را که مهار کننده سیستم ایمنی است تولید کنند و مانع تولید طوفان سایتوکاینی شوند. بنا براین هم ماکروفاژها هم لنفوسیت ها و هم نوتروفیل ها همه در این طوفان نقش دارند.
وقتی که سلول های پنوموسیتی با ویروس کرونا عفونی می شوند باعث می شوند که NF-KB شروع به تولید سایتوکاین ها از جمله اینترلوکین 6 بکنند. اینترلوکین 6 یک کموکاین بسیار قوی در تولید طوفان اینترلوکینی است. اینترلوکین 6 باعث حفاظت از عفونت می شود در جلوگیری از صدمه بافتی دخالت می کند اما تولید زیاد آن موجب دردسر می شود. اینترلوکین شش به رسپتورهای خود می چسبد و زیر هپاتوسیل ها لنفوسید ها منوسید ها بی سل ها تی سل ها MKCELL ها ماکروفاژها نوتروفیل ها در پی آن اینترلوکین بیشتری تولید می شود. MCP1 , EEGF و کاهش E – CADHERIN می شود. ترکیب اینترلوکین 6 با رسپتورش بر روی سلول های دندرتیک باعث تولید TH17 می شود. ظهور سایتوکاین های التهابی درست بعد از پیکی است که بعد از یک افت دیده می شود به جز اینترلوکین 1 و رسپتور آن. این نشان می دهد که اینترلوکین 1 ممکن است در ظهور کرونا در مراحل ابتدایی نقش مهمی داشته باشد به علاوه NETS- IL1β در بیماران شدید کوویدی دیده می شوند. اینترلوکین 1 و تی ان اف موجب تولید یالورونان می شوند که یک فاکتور مهم در تولید ARDS در کروناست.بنابراین اینترلوکین 1 در شروع بیماری کرونا نقش مهمی می تواند بازی کند.
برعکس تیپ 1 اینترفرون شامل اینترفرون آلفا و بتا و تیپ 3 اینترفرون (لامبدا ) در بیماران شدید کرونایی اختلال شدیدی پیدا می کند این اینترفرون ها نقش مهمی در ایمنی ضد ویروسی دارند. در نتیجه این نقص اینترفرون ها ما شاهد گسترش ویروس کرونا هستیم. مطالعاتی نشان داده است که تجویز اینترفرون تایپ 1 در ابتدای بیماری می تواند مفید باشد.به علاوه بعد از عفونت ویروسی اینترفرون تایپ 1 مهار می شود. ما می توانیم به این نتیجه برسیم که ایمنی نقش بسیار مهمی در بیماران کرونا دارد. گزارش شده است که بیماران شدید کرونا ممکن است با مرگ و میر 61 درصدی همراه باشند.
درمان کرونا:
آنتی بادی های خنثی کننده باید طوری عمل کنند که S پروتئین به رسپتور آن یا در واقع RBD DOMAIN وصل نشود.متاسفانه اخیرا آنتی بادی هایی که محل اتصال ACE2 را مورد هدف قرار می دهند نا توان از این هستند که S1 یونیت ویروس به رسپتورها وصل شود.تنها دو تا از سه تا دومین های RBD می توانند مانع اتصال ویروس به غشا شوند. اثربخشی آنتی بادی های بی اثرکننده وابسته است به قدرت ACE2 برای RBD DOMAIN .
دومین درمان درمان پلاسمای بیماران بهبود یافته است. به هر حال این درمان هنوز نیاز به بررسی های بیشتری دارد.یکی از درمان های جدید مهار کننده رسپتور اینترلوکین یک بتا است به نام آنا کینرا و کاناکینومب. در حال حاضر نتایج این داروها کاملا مشخص نیست و یکی از تحقیقات این دارو را در بیماران ملایم تا متوسط استفاده کرده است که این خود یک مشکل اساسی در آن تحقیق است باید دارو را در مراحل شدید استفاده کرد تا بتوان دریافت که آیا این داروها موثر هستند یانه . یکی دیگر از داروها اماپالومب می باشد که آنتی بادی است بر ضد اینترفرون گاما این دارو نیز همانند داروی مهار کننده GM-CSF در حال مطالعه است. داروی بعدی اینترفرون یک و اینترفرون 3 می باشد. اینترفرون آلفا دو بی به تنهایی یا در اشتراک با آر بی دول توانسته اند طول مدت ویروس های قابل شناسایی را و همچنین مارکر های التهابی را کاهش دهند.یک مطالعه از ترکیب ریباورین لوپیناویر – ریتانویر و اینترفرون بتا یک بی در 127 بیمار کرونایی استفاده شد. که باعث شد میزان ماندن بیماران در بیمارستان کاهش پیدا کند. یکی دیگر از راه ها تعویض پلاسما هست. در حال حاضر دو مطالعه بر روی این مسئله انجام می گیرد که به همراه روکسولیتینیب می باشد.درمان دیگر تزریق IGG است.درمان پیشنهادی بین 0.3 تا 0.5 گرم به ازای هر کیلو به مدت 5 روز متوالی. در حال حاضر این هم در حال مطالعه می باشد.