پاتوژنسیس و درمان IPF :
فیبروز ریه یک بیماری پیشرونده است و در نهایت منجر به نارسایی تنفسی با افزایش ECMD می شود. شروع فیبروز با یک تحریک متوالی شروع می شود و منجر به ترمیم زخمی می شود که ناهنجار است به همراه عملکرد بد فیبروبلاست ها که در نهایت به فیبروز انتهایی تبدیل می شود اگرچه علت فیبروز مشخص نیست اما ایمنی ذاتی و انطباقی در آن نقش دارد. اهمیت التهاب به عنوان یک پایه ای از فیبروز غیر قابل قطعی است و مورد بحث قرار دارد. همچنین رول های جدیدی در ماکروفاژها از دست رفتن تحمل B و C سل ها دیده می شود که در نهایت منجر به پاسخ فیبروزی می شود. فیبروز ریه بدون درمان دو تا سه سال عمر می کنند. در 2014 پرفنیدون و NINTEDANIB(OFEV) برای درمان فیبروز و آهسته کردن روند آن تصویب شد اگرچه این درمان ها پیش آگهی بهتری برای بیماران پیش آوردن اما فیبروز یک بیماری غیر قابل درمان است بدون اینکه علتش به شکل واقعی شناخته شده باشد.
از نظر هیستولوژیک فیبروز به لحاظ تجمع زیاد از حد پروتئین های ماتریکس خارج سلولی یا ECM حضور فوکوس های فیبروبلاستی و مناطق فیبروتیک در مجاورت با ریه سالم که به اصطلاح فیبروز هتروژنوس از نقطه نظر فضایی گفته می شود. اساس التهاب بیش از حد در فیبروز یکی از مسائلی است که شاید در سیگنال های TGFβ نهفته باشد.
برخی بیماری های بافت نرم مثل اسکلوروز سیستمیک رماتیسم مفصلی همراه هستند با ILD شامل فیبروز ریه. همچنین ژن ها هم در تشکیل فیبروز خانوادگی نقش داشتند. به عنوان مثال تجربه نویسنده حاکی از سه فیبروز در یک خانواده بود که همه آنها قبل از 50 سالگی فوت کردند. تغییرات ژنتیکی در پروتئین های واکنش دهنده به TOLL به نام TOLLIP موجب مهار پروتئین TLR می شود که در نهایت موجب می شود به افزایش پاسخ التهابی در بیماران فیبروزی. به علاوه نقص عملکرد TLR3 همراه است با یک نوع شدید فیبروزی.
از آنجایی که این بیماری در افراد مسن بیشتر دیده می شود پدیده ایمنی سنسنس ممکن است نقشی اساسی در این موضوع بازی کند. در واقع تغییرات TLR ممکن است منجر به این پدیده شود و کاهش فعالیت TLR در فیبروز فامیلی دیده می شود.
علی رغم اینکه تئوری های زیادی در مورد التهاب در فیبروز وجود دارد اما کورتن ها نتوانسته اند در درمان فیبروز موثر واقع شوند. بنابراین اول باید دید که التهاب تا چه سطحی در به وجود آمدن مریضی دخالت دارد ما در اینجا صحبت از پلیس خوب و بد می کنیم. ما در این مقاله سغی می کنیم که ایمنی ذاتی و انطباقی را در ایجاد فیبروز مورد بررسی قرار دهیم.
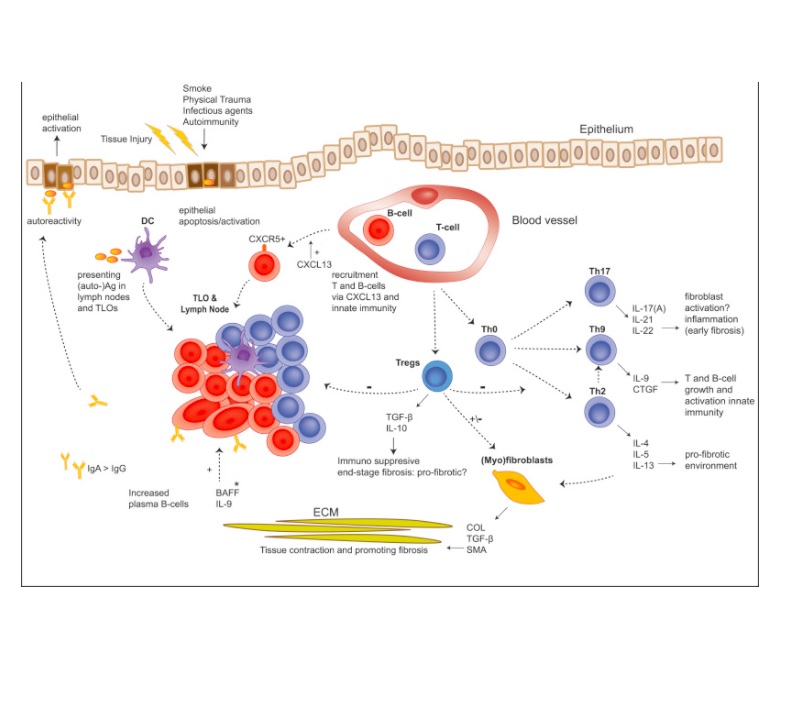
نوتروفیل ها: هنگامی که یک ضربه به ریه وارد می شود نوتروفیل ها اولین پاسخ دهنده ها به خصوص نسبت به IL8 که باعث تجمع نوتروفیل ها می شود هستند. نوتروفیل ها رسپتورهای دیگری هم مثل GM-CSF و G-CSF دارد.
نوتروفیل ها سایتوکاین های التهابی و ROS ترشح می کنند. در لاواژ بیماران فیبروزی IL8 و G-CSF زیاد می شود.وجود نوتروفیل ها در لاواژ بیماران با ارزش FVC تناسب معکوسی دارند و در صورت وجود IL8 می توان حدس زد که بیماران با حملاتی در آینده مواجه خواهند شد. در موش مهار IL8 باعث کاهش فیبروز ناشی از بلئومایسین می شود. التهاب یا ارتشاح نوتروفیلی در لاواژ بیماران ممکن است ناشی از وسعت بیماری باشد و هیچ ارتباطی بین آن و آینده بد بیماری وجود ندارد. باید توجه کرد که فیبروز ناشی از بلئومایسین در موش ها یک بیماری پیش رونده برخلاف فیبروز انسانی نیست و بسیار وابسته به التهاب می باشد. یکی از تولیدات نوتروفیل ها الاستاز نوتروفیلی است (NE ) که هم خصوصیت ضد فیبروتیکی و هم خصوصیت فیبروتیکی دارد. NE موجب می شود که کلاژن 4 و الاستین تخریب شود و در نتیجه فیبروز را بهبود می بخشد. برعکس LE می تواند موجب ارتشاح فیبروبلاستی شود و موجب پیشرفت مایوفیبروبلاستی شده و موجب ترشح TGFβ بشود که همه اینها موجب افزایش قیبروز می شود . دو مطالعه در زمینه مهار کننده های NE نشان داد که فیبروز در آسبستوز و مدل فیبروز بلئومایسینی بهتر شود.
نوتروفیل ها نقش مهمی را در فاز شدید التهابی دارند و می توانند موجب تغییر ساختمانی ریه و فیبروز شوند بنابراین نقش نوتروفیل ها در فیبروز هنوز ناشناخته می باشد.
ماکروفاژها:
ماکروفاژها سلول های با خصوصیت پلاستیک هستند و می توانند از شکل M1 به M2 یا برعکس تغییر جهت دهند. ماکروفاژهای M1 ماکروفاژهایی هستند که یه صورت کلسیکالی به وسیله LPS و اینترفرون گاما فعال می شوند و سایتوکاین های التهابی مثل TNFα و IL1 و IL6 ترشح کنند. ماکروفاژهای M2 که در واقع ماکروفاژهای آلترناتیو نام دارند که در تماس با اینترلوکین 4 – 10 و 13 تحریک می شوند و خصوصیت ضد التهابی دارند. و در واقع خصوصیت فیبروتیک دارند. در شذایط التهابی مزمن ماکروفاژهای M1 به تدریج به ماکروفاژهای M2 تبدیل می شوند که بتوانند زخم ها را مداوا کنند. همانطور که انتظار می رود ماکروفاژها ی M2 در ریه های فیبروتیک وجود دارد و فاکتورهای رشد زیادی مثل TGF-β فیبروبلاست گروتفکتر (FGF) ، PDGFα ، IGF1 و VEGF ترشح می کنند. VEGF همراه است با شدت بیماری در بیماران IPF ماکروفاژهای M2 در تشکیل ماتریکس های خارج سلولی هم نقش دارند به خصوص در تولید کلاژن توسط افزایش متابولیسم آرجینین
بیماران فیبر.زی که مبتلا به حملات بیماری می شوند میزان CCL18 آنها در لاواژ زیاد می شود که بیشتر از همه توسط ماکروفاژهای M2 ترشح می شود و همراه است با حملات بیشتر فیبروزی. این نشان می دهد که ماکروفاژها در حملات فیبروزی نقش دارند. CCL18 در بیماران فیبروزی استیبل هم در لاواژ ریاد می شود. CCL18 موجب جذب تی سل ها به ریه می شود و فیبروبلاست ها را فعال می کند. در عوض فیبرو بلاست ها و کلاژن ماکروفاژها را تحریک می کنند تا CCL18 تولید کنند در انسان یک نمونه از ماکروفاژهای M2 وجود دارد که به نام ماکروفاژهای لگولاتوری نام برده می شود و این ماکروفاژها می توانند ذرات ECM را با ترشح MMP از بین ببرند و موجب کاهش فیبروز بشوندو رول MMP ها و مهار کننده های آنها در جای دیگر مورد بحث قرار گرفته استو بنا براین ماکروفاژها ممکن است خصوصیت ضد فیبروتیکی – فیبروتیکی و عملکرد بافتی رجرانتیو داشته باشند. ماکروفاژهای M2 نقش مهمی را در تنظیم فیبروز بازی می کنند و حتی در حملات فیبروز هم تقش دارند.
منوسیت ها:
منوسیدهی انسانی در هموستاز بافتی و ایمنی در واقع تولید کننده ماکروفاژها هستند. CCL2 یکی از مهم ترین کموکاین هایی هست که باعث مهاجرت منوسیدها می شود و به MCP1 معروف است CCL2 به CCR2 وصل می شوددر بیماران فیبروزی میزان CCL2 و افزایش تولید آن توسط سلول های آندوتلیال مشاهده شده است که نشان می دهد منوسیدها به سمت ریه بیمران فیبروزی مهاجرت می کنند. یک دارو به نام کارلومب وجود دارد که در فاز دو یک مطالعه است و CCL2 را مهار می کند اما در بیماران IPF نتوانسته است میزان CCL2 را کاهش دهد. منوسیدها در رده بندی های مختلفی وجود دارند شامل منوسید های کلسیکال با اکسپرسن CD14HI منوسیدهای بینابینی با CD14HI , CD16HI و منوسیدهای غیر کلسیکال با CD16HI مشخص می شوند اگر چه نقش واقعی این انواع در IPF نامشخص می باشد اما ارتشاح منوسیدهای کلسیکال در بیماران فیبروتیکی جایی که آنها به ماکروفاژ تبدیل می شوند دیده می شود. افزایش منوسیدهای کلسیکال و بینابینی همراه است با پیشرفت بیماری و پروگنوز بد بیماری. منوسیدهای بینابینی تولید اینترفرون آلفا CCL3 یا MIB-α و CCL4 یا MIP-1β که همه اینها باعث تمایز مایوفیبروبلاستی می شوند. بعد از فعال شدن منوسیدهای کلسیکال و بینابینی از بیماران SS تولید بیشتری CCL18 و اینترلوکین 10 می کنند. در بیماران SS که فیبروز ریه دارند این منوسیدها با CD14+ باعث تولید اینترلوکین 6 – تی ان اف آلف و تی جی اف بتا نسبت به آنهایی که فیبروز ندارند تولید می کنند.این نشان می دهد که التهاب مزمن می تواند منوسیدها را به سمت شکل فیبروتیکی تغییر دهد. نقش منوسیدهای غیر کلسیکال در بیماران فیبروزی نامشخص است. در بیماران فیبروزی مبتلا به SS افزایش منوسیدهای CD16+ همراه با شدت فیبروز ریوی . ناشر این مقاله پیشنهاد می کند که منوسید های CD16+ می تواند منشا اصلی فیبروتیکی ماکروفاژها باشد در خلاصه عملکرد منوسیدها در جریان فیبروز به نظر می رسد فیبروتیکی باشد و باعث تولید سایتوکاین های التهابی پروفیبروتیکی باشد.
فیبروسید ها:
فیبروسیدها از منوسیدها نشات می گیرند و فیبروبلاست ها را تشکیل می دهند. فیبورسیدها در پاسخ به فاکتورهای کموکاینی وارد محل زخم می شوند وسلول های شبیه فیبروبلاست ها را تشکیل می دهند که قسمت کوچکی از زخیره فیبروبلاست هارا تشکیل می دهند. فیبروسیدها CD45 اکسپرس می کنند به اضافه کلاژن یک داخل سلولی به علاوه رسپتورهای CXR4 ، CCR7 , CCR2 برای شناخت فیبروسید ها استفاده می شوند. به عنوان مثال فیبروسیدهایی که CCR7 مثبت هستند مقادیر زیادی TGF-β تولید می کنند برای کمک به ترمیم زخم پروتئین های زیادی شامل کلاژن 1 کلاژن 3 VIMNTIN و فیبرونکتین توسط فیبروبلاست ها تولید می شود علاوه بر آن فیبروبلاست ها قدرت تولید IL8 , IL6 , TGFβ ,PDGF, GMCSF, VEGF می باشند.این فاکتورها باعث تمایز فیبروبلاست ها می شوند تولید عروق جدید می کنند و سلول های التهابی را به منطقه فرا می خوانندو فیبروسیدها همچنین باعث تولید کلاژن تایپ 1 MMP1 و PDGFβ توسط فیبروبلاست ها می شوند.
فیبروبلاست ها به ریه مهاجرت می کنند که تحت تاثیر CXCL12 که به رسپتور CXCR4 می چسبند هستند.
افزایش مقدار cxcl12 در بیماران فیبروژی در لاواژشان دیده می شود و با TLCO هماهنگی دارد. دارویی که CXCL12 را خنثی می کند می تواند فیبروز ریه در موش ها را کم کند. در مهاجرت فیبروسیت ها به ریه CXCL12 نقش مهمی بازی می کند و مقدار فیبروسیت در خون با پیش آگهی بیماری ارتباط دارند. در این مطالعه نشان داده شد که اگر 50 درصد از لوکوسید ها فیبروسید باشند نشان می دهد که بیماری به زودی به مرگ منتهی می شود. اکثر بیماران با بیش از 5 درصد فیبروسید به حملات فیبروزی مبتلا می شوند.
نوع دوم سلول های لنفوئیدی و ماست سل ها:
این مشخص شده است که سایتوکاین های تی هلپر 2 در فیبروز ریوی نقش دارند IL4,5,9,13 به عنوان سایتوکاین های تایپ 2 ILC2 معرفی شده اند. سایتوکاین هایی که ILC2 را به ریه می آورند IL25,IL33 هستند که توسط ضربه به اپیتلیوم و ماست سل های فعال شده و ائوزینوفیل ها و ماکروفاژها ترشح می شوند. همه این سلول ها نقشی در فیبروز بازی می کنند. در واقع هم تعداد ILC2 و اینترلوکین 25 در لاواژ بیماران فیبروز ریوی زیاد می شود. IL13 توسط ILC2 ترشح می یابد و نقش مهمی در فیبروز ریوی دارد. IL13 منجر به فعالیت استت 6 می شود که به نوبه خود تایپ 2 ایمنی را و سنتز کلاژن را کنترل می کند. علاوه به آن اینترلوکین 13 یک ترشح کننده قوی TGFβ می باشد. اخیرا سه مطالعه روی مهار کننده های اینترلوکین 13 و 4 نتوانسته اند نتایج امیدوارکننده ای ایجاد کنند. در فیبروز بیماران SS تعداد ILC2 زیاد می شود و با شدت فیبروز هماهنگی دارد. IL9 می تواند ILC2 را تحریک کند و مهار IL9 موجب مهار راه های هوایی و تا قسمتی کاهش TGF-β شود. IL2 همچنین می تواند آمفی رگولین ترشح کند . آمفی رگولین موجب افزایش سلول های فیبروبلاست می شود و تولید کلاژن را افزایش می دهد. در مورد ILC1 که تولید اینترفرون گاما و TNFα می کند و ILC3 که تولید اینترلوکین 17A و تولید اینترلوکین 22 می کند مطالعات بیشتری لازم می باشد.
مست سل ها حاوی گرنول هایی هستند که شامل اینترلوکین 4 و 10 و 13 هستند به اضافه TGFβ , PDGF افزایش تعداد ماست سل ها در ریه و افزایش تولیدات مست سل ها در ریه شامل تریپتاز کیماز و هیستامین در لاواژ بیماران فیبروزی دیده شده است. هیستامین موجب افزایش فیبروبلاست ها می شود و تریپتاز موجب افزایش تایپ 1 کلاژن از فیبروبلاست ها می شود. فعلا مطالعات روی موضوع ILC2 و ماست سل ها نیاز به بررسی بیشتری دارند.
دندرتیک سل ها هم نقشی در ایمنی ذاتی و هم نقشی در ایمنی انطباقی دارند. دندرتیک سل ها در ایجاد ساختمان های لنفوئید یا TLO در فیبروز دیده می شوند. TLO ها ساختمان مشابهی با لنف نود ها دارند و شامل لایه های تی سل ها هستند با فولیکول های بی سل همچنین آنها ونول ها پر از ونول ها هستند ولی کپسول ندارند. دندرتیک های نابالغ در نواحی فیبروز دیده می شوند و دندرتیک های بالغ در TLO ها دیده می شوند. دندرتیک ها نقش موثری در حفاظت از TLO ها بازی می کنند. TLO ها در واقع به علت التهاب های مزمن در محل های لوکال ایجاد می شوند و ممکن است با افزایش فیبروز گسترش پیدا کنند یکی از کموکاین های موثر برای جذب دندرتیک ها از طریق رسپتور CCR می باشد که با CLL19 که از سلول های اپیتلیال و فیبروبلاست ها ترشح می شوند تحریک می شوند. CCL19 در لاواژ بیماران فیبروزی همراه با دندرتیک های نا بالغ دیده می شوند جالب اینست که فیبروبلاست ها می توانند فعالیت دندرتیک ها را مهار کنند. اما این به علت شرایط سلول های ضد التهابی استرومال هست تا کاهش فعالیت دندرتیک ها. به علت فعالیت ضد ایمنی سلول های استرومال می باشد تا به علت کاهش فعالیت دندرتیک ها. مطالعات نشان می دهد که دندرتیک ها در التهاب فیبروز ریه نقش دارند
تی سل ها نقش در فیبروز ریه دارند مانند تشکیل ECM و افزایش فیبروبلاست ها یک عدم تعادل بین TH1/TH2به نظر می رسد در مرکز ایجاد فیبروز ریه نقش داشته باشد. در واقع تایپ 2 سایتوکاین ها فیبروتیک هستند و تایپ 1 سایتوکاین ها مثل اینترفرون گاما و اینترلوکین 12 نقش محافظتی دارند اما مطالعات به نام INSPIRE نتوانست تاثیر اینترلوکین گاما را بر روی IPF نشان بدهد. در گذشته شواهدی در نقش TH17 / TH9 و TREG ها تصور می شد ما بر روی TH9 , TH17 , TREG ها بحث می کنیم اول TH9 ، TH9یک تی هلپر جدیدی است که بر روی سلول های سرطانی با فعالیت ضد انگلی و بیماری های اتو ایمون می تواند موجب فعالیت التهاب آلرژیک شود. IL4 و THFβ در سایتوکاین هایی هستند که با فیبروز نقش دارند و همچنین با تایپ 2 ایمنی و باعث می شوند که تمایز TH9 ایجاد شود. TH9 به نوبه خود IL9 را تولید می کند و IL9 موجب افزایش تی سل بی سل CCL11 و اینترلوکین 8 از سلول های صاف می شوند. و باعث فعالیت ILC2 می شوند. IL9 هم فعالیت فیبروتیکی و هم فعالیت ضد فیبروتیکی دارد. در واقع مهار IL9 در موش ها باعث بهبود فیبروز کبدی شده است. مهار IL9 باعث کاهش TH17 می شود به همراه TH1 و باعث کاهش اینترلوکین 17 A و اینترفرون گاما TGFβ اینترلوکین 6 و TNFα می شود. در مطالعه ای نشان داد که اینترلوکین 9 در موش ها باعث افزایش فیبروز ریوی می شود که احتمالا به خاطر افزایش ائوزینوفیل ها و CTGF می باشد. در واقع یک اثر متضاد بین TH9 و اینترلوکین 9 در فیبروز دیده شده است و در حال حاضر نمی توان به راحتی در مورد آن تصمیم گیری کرد.
TH17 موجب تولید IL17A اینترلوکین 21و22 می شود و در بسیاری از بیماری های اتوایمون نقش دارد و بر ضد باکتری های خارج سلولی و قارچی نقش دارد. امروزه یک افزایش فعالیت TH17 در بیماری های مرتبط با خود ایمنی به نام COPA و ارتباطش با TH17 و فیبروز ریه کشف شده است. TH17 و IL17 در بیماران فیبروزی اطراف التهاب ها در بیماران فیبروزی وجود دارد و مشخص است که اینترلوکین 17 یک نقشی در فیبروز ریه بازی می کند. باید توجه کرد که اینترلوکین 17 فقط توسط TH17 ترشح نمی یابد و سایر سلول ها مثل ماکروفاژها نوتروفیل ها NKCELL ها ILC3 دلتا گاما تی در تولید اینترلوکین 17 نقش دارند. اینترلوکین 17 موجب افزایش کلاژن TGFβ و اینترلوکین 6 می شود. آنتی بادی بر ضد اینترلوکین 17A موجب کاهش فیبروز در بلئومایسین و مدل های رادیوتراپی شده است. به علاوه اینترلوکین 17 سلول های اپیتلیال را فعال می کند باعث تولید اینترلوکین 8 می شود که باعث جذب نوتروفیل ها می شود. TH17همچنین باعث افزایش التهاب توسط کاهش TREGها می شود در واقع مهار اینترلوکین 17 باعث می شود ک نوتروفیل ها کمتر تجمع پیدا کند و فیبروز بهبود پیدا کند و عملکرد TREG ها بهبود یابد افزایش TREG ها بیشتر از همه توسط کاهش اینترلوکین 6 حادث می شود. همچنین افزایش عملکرد اینترلوکین 10 هم باعث افزایش فعالیت TREG ها می شود افزایش اینترلوکین 10 به همراه TGFβ موجب بهبود TREG ها نسبت به TH17 می شود. به علاوه شیفت بین TREG ها و TH17 به وسیله اینترلوکین 27 موجب بهبود فیبروز در مدل بلئومایسینی می شود.بنابراین یک تعادل بین فعالیت ضد التهابی TREG ها و فعالیت TH17 در توسعه فیبروز ریه وجود دارد. جالب توجه این است که TH17 و IL17A برای التهاب اولیه و IL17A برای توسعه فیبروز ریه در مدل سیلیکوز لازم هستند در انسان ها فیبروز پوستی سیگنال IL17 می تواند خصوصیت ضد فیبروتیکی داشته باشد. نقش واقعی TH17 و اینترلوکین 17 در توسعه فیبروز ریه کاملا شناخته شده نیست.
اگر چه اکثر مطالعات بر روی موش ها نشان می دهد که TH17 و IL17 نقش اولیه ای در شروع فیبروز دارند اما در انتهای فیبروز ممکن است نقش بارزی نداشته باشند.
TREG ها : موجب تولید TGFβ و تولید فیبروبلاست ها و PDGFβ می شود یکی از اولین مطالعات روی TREG ها در بیمارن فیبروزی نشان داد که TREG ها در لاواژ و خون محیطی بیماران فیبروزی کاهش عملکرد داشتند . کاهش عملکرد TREG ها با کاهش FVC و TLCO ارتباط دارند. موش هایی که TREG ها در ریه هاشان زیاد هست کاهش فیبروز را نشان می دهند بنابراین مطالعات اولیه نشان می دهد که TREG ها فعالیت های ضد فیبروتیکی دارند. اما مطالعات بعدی نشان داد که کاهش TREG ها در موش ها باعث افزایش فیبروسید ها و افزایش فیبروز شده است. تزریق TREG ها باعث تشدید فیبروز بلئومایسینی می شود در نتیجه نویسنده مقاله به این نتیجه رسید که وجود TREG ها نقش نامساعدی در تولید تایپ 2 ایمنی دارند و باعث افزایش التهاب می شوند اخیرا نشان داده شده که فعالیت TREG ها در بیماران فیبروزی زیادتر شده است.اینکه ما در روی فاز اولیه یا آخر فیبروز بحث بکنیم مهم است. TREG ها ممکن است در اوایل بیماری مضر باشند ولی در اواخر بیماری فیبروز در موش ها سودمند باشند.از نظر بافت شناسی تصور می شود که TREG ها نقش محافظتی در فیبروز داشته باشند اما مطالعات بیشتری در این زمینه نیاز می باشد.
بی سل ها در TLO ها وجود دارند این ها نقشی در تولید اتو آنتی بادی های دارند بی سل های رسپتورهای مختلفی دارند برای CXCL13 و CXCR5 و CR6 و CR7 . CXCL13 روی رسپتور CXCR5 اثر می کند که خود توسط دندرتیک های فولیکولار و فولیکولار تی هلپرها تولید می شود. CXCL13 در بیماری های فیبروزی افزایش می یابد و این افزایش مخالف پیش آگهی بیمار فیبروزی است. در بیماران فیبروزی فاکتور فعال کننده بی سل به نام BAFF با توسعه بیماری ارتباط دارد اتو آنتی بادی های تولید شده توسط بی سل ها آنتی بادی های خودی را مورد هدف قرار می دهند و موجب شناخت آنتی ژن های اپیتلیال می شوند این ها باعث ایجاد التهاب و ترمیم نابه جای زخم می شود. میزان این آنتی بادی ها با پیش آگهی بیمار ارتباط دارد برای مثال بیمارانی که فیروز دارند و مقدار IGA آنها بیشتر از 2.85 گرم در لیتر باشد پیش آگهی بدی دارد. این نباید موجب تعجب باشد چون که افزایش TGFβ در بیماران فیبروزی زیاد می شود. به علاوه آنتی بادی هایی که بر ضد برخی آنتی ژن ها هستند از جنس IGA هستند.
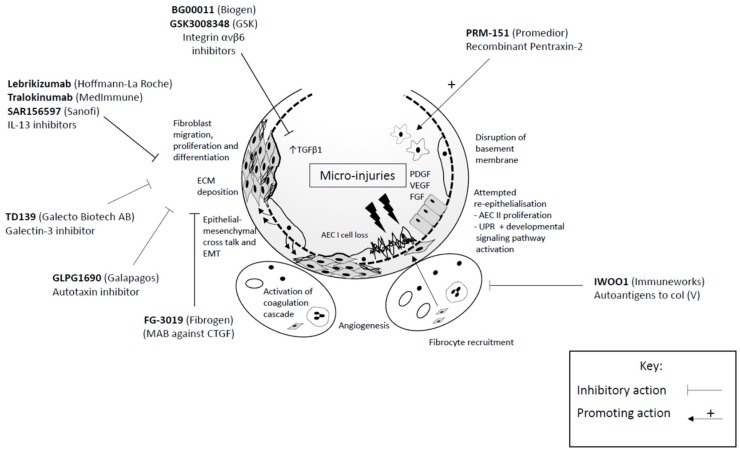
در سال 2014 پرفنیدون و نینتدانیب برای درمان فیبروز به تصویب رسید. پرفنیدون یک دارویی است که در ابتدا برای مقاصد ضد درد و ضد تب کشف شد. یکی از مهم ترین آثار پرفنیدون تاثیر روی TGFβ است. پرفنیدون باعث کاهش TGFβ می شود که خود موجب کاهش فیبروبلاست ها می شود. تولید کلاژن به وسیله پرفنیدون و نینپدانیب کاهش پیدا می کند.و باعث می شود کلاژن های خارج سلولی به صورت فیبریل کاهش یابد. پرفنیدون باعث کاهش سایتوکاین های التهابی می شود و موجب کاهش TH2 می شود. بیمارانی که پرفنیدون مصرف می کنند میزان سایتوکاین های TH2 آنها کاهش پیدا می کنند. داروی بعدی نینپدانیب می باشد که باعث می شود رسپتورهای تیروکایناز شامل PDGFR – FGFR – VEGFR کاهش پیدا کند خصوصیت ضد التهابی این دارو کمتر شناخته شده است در فرم فیبروزی بلئومایسین و CDK این دارو موجب کاهش ارتشاح نوتروفیل ها و منوسیدها به خصوص وقتی که در دوز پروفیلاکتیک داده می شوند می شود.خصوصیت ضد التهابی این دارو وقتی که در دوز های بالا داده می شود مثلا 10 میلی گرم یا بیشتر نامشخص است. سوم داروهایی که در حال مطالعه هستند یکی از آنها داروی آنتی TNFα اینترفرون گاما می باشد. هیچکدام از این داروها تا کنون اثربخشی نشان ندادند.شاید علت این موضوع این باشد که فیبروزهایی که در این مدل ها استفاده شده اند فیبروز های بلئومایسینی باشند. نکته ی دیگر این است که علت حملات فیبروز از فیبروز پیش رونده کند متفاوت است این به این معناست که داروهای جدید برای درمان فیبروزهای استیبل ممکن است در درمان حملات فیبروزی موثر نباشند. یکی دیگر از این داروها پنتراکسین 2 می باشد که موجب مهار منوسیدها به سمت ماکروفاژها و فیبروسیدها می شوند. نتایج اولیه این دارو نشان دهنده یک بهبود کوچک ولی مهم در راه رفتن شش دقیقه ای و FVC شده است.
آنتی بادی که سایتوکاین اینترلوکین 13 را مهر می کند در فاز دوم قرار دارد. و تا کنون نتوانسته است سودمندی خود را نشان دهد . آنتی بادی ضد اینترلوکین 4 و 13 هم نتوانست سودمندی خود را نشان بدهد. البته داروی ضد اینترلوکین 13 نشانه هایی از مثبت بودن را در حملات فیبروزی نشان داد این نشان می دهد که تایپ 2 ایمنی ممکن است نقشی در حملات فیبروزی داشته باشد. یکی دیگر از داروها به نام پم رو لومب می باشد که باعث مهار CTGF شده و باعث کاهش اینترلوکین 1 بتا و CCL3 می شود و در نهایت باعث کاهش تجمع ECM می شود فاز 2 این دارو پروفایل سالمی را از خود نشان داده است. و باعث افزایش FBC بعد از 48 هفته از درمان شده است. داروی بعدی ریتوکسی مب می باشد که یک دارویی است بر ضد CD20 و در درمان بیماری های اتو ایمون استفاده می شود این دارو موجب بهبود علائم بیمار و TLCO بیمار در حملات فیبروزی شد. فاز 2 مطالعه روی این دارو در بیماران فیبروزی استیبل در حال صورت گرفتن می باشد. داروی دیگر دارویی است بر خلاف مهار کننده BAFF که باعث می شود بی سل ها را مهار کند این دارو در حال حاضر در حال مطالعه است.
خلاصه اینکه داروهای جدید مثل GLPG1690 و PMR151 در فازهای دوم به سر می برد. باید توجه داشت که التهاب در ریه برای مهار عفونت اهمیت دارد. بنابراین باید تعادلی بین التهاب و از بین بردن التهاب وجود داشته باشد.
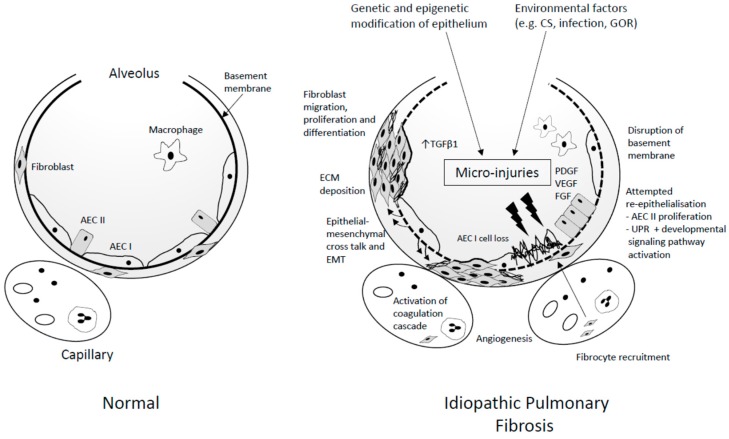
در واقع فیبروز ریه از نظر هیستولوژیک همان UIP می باشد که توسط HRCT یا بیوبسی ریه تشخیص داده می شود. تشخیص قطعی UIP می تواند بر اساس HRCT انجام بشود اگر کیست های زنبورعسلی و رتیکولیشن های غالب زیر پرده پلور وجود داشته باشد بدون اینکه برونشکتازی یا برونشیولکتازی دیده شود. از آنجایی که گرفتن نمونه جراحی به خصوص در افراد پیر می تواند منجر به مرگ و میر شود پیشنهاد می شود که تشخیص احتمال UIP در HRCT به همان اندازه بیوبسی ارزش دارد. بنابراین برخی از محققان پاسیب UIP را با احتمال UIP جایگزین کرده اند.د تا بتوانند حضور رتیکولیشن و برونشکتازی و برونشیولکتازی را در پایه و زیر پلور ارائه دهند. اینها اعتقاد دارند که با این روش می توان نیاز به جراحی را گرفت.
فیبروز از نقطه نظر بافت شناسی هم از لحاظ زمانی و هم از نظر فضایی هتروجنوس می باشد. و تجمعات فیبروبلاست و میوفیبروبلاست همراه با تجمع نا منظم کلاژن و ECM باعث می شود که ترکیب بافتی ریه بهم بخورد و کیست های لانه زنبوری تشکیل شود. در حال حاضر تئوری غالب بر این است که فیبروز ناشی از توروماهای مکرر ریه حادث می شود. موضوع اول با توجه به اینکه فیبروز در برخی از اعضای خانواده دیده می شود مسئله تغییرات ژنتیکی است سورفاکتانت توسط پونوموسیدهای نوع دوم برای حفاظت از آلوئول ها تولید می شود. انواعی از سورفاکتانت مثل SP-C و SP-A2 در فیبروزهای خانوادگی نقش دارند. این نوع سوزفاکتانت ها در نهایت باعث می شوند که سلول های پوششی آلوئول از بین برود تغییرات پروتئینی در پونوموسیدهای نوع دوم دیده شده است. گرچه این تغییرات در تولید فیبروز در موش ها به مدت شش ماه نتوانسته است فیبروز ایجاد کند.
از طرف دیگر MUC5B توانسته است در فیبروزهای خانوادگی کشف شود. این جهش ژنتیکی در 20 درصد از فیبروزهای خانوادگی دیده می شود. نقش این جهش های ژنتیکی در ایجاد تولید زیاد موکوس و پاک کردن آنها در راه های هوایی باعث می شود که التهاب مزمن ایجاد شود. MUC5B نه تنها پیش آگهی فیبروز را پیش بینی می کند بلکه میزان مرگ و میر را هم دو برابر می کند.
موتاسیون دیگری که در فیبروز اتفاق می افتد به آن TOLLIP می گویند.این موتاسیون هم همراه است با افزایش مرگ و میر در بیماران فیبروزی. مسئله دیگر تلومراز می باشد. باعث می شود کروموزم ها از تخریب جلوگیری شود. نقص این آنزیم ها در طول مسن شدن کاهش پیدا می کند و باعث می شود که کروموزوم ها در معرض تخریب قرار بگیرد. چندین جهش در این آنزیم ها وجود دارد که شایع ترین آن ها TERT می باشد. این موتیشن ها در فیبروزهای فامیلی دیده می شوند و مواردی از سایر بیماری های بافتی هم با این موتیشن ها ارتباط دارند. موتیشن ها همراه هستند با پیش آگهی بد بیماری. در حال حاضر مطالعاتی بر روی این پدیده سنسن در حال مطالعه است. جلوگیری از این پدیده می تواند موجب بهبود عملکرد ریوی و فیبروز شود. دارویی به نام ناویتوکلکس که یک مهار کننده BCL2 می باشد تنوانسته است جلوی فیبروز ریوی را در مدل پرتو درمانی بگیرد.
عوامل محیطی از میان عوامل محیطی شامل گرد و خاک های فلزی آلودگی رفلاکس سیگار و عفونت سیگار مهم ترین علت ایجاد فیبروز می باشند. و شواهدی نشان می دهد که با قطع سیگار بهبودی در بیماری ایجاد می شوند.سیگار باعث تغییراتی در DNA می شود که مستعد کننده فیبروز می باشد. از میان ویروس ها HHB بیشترین مطالعه را به خود اختصاص داده است و در میان فیبروزی ها میزان ابتلا به این ویروس 97 درصد گزارش شده است. ویروس دیگر EBV می باشد. مسئله دیگر وجود باکتری هاست که نقش آنها به خصوص در حملات فیبروز مطرح شده است. در مطالعه ای در چین به بیماران فیبروزی 12 ماه کوتریماکسازول داده شد. اگرچه این دارو موجب تغییری در FVC نکرد اما موجب کاهش عفونت و مرگ و میر شد. مطالعه ی دیگری در حال انجام می باشد. مسئله دیگر ریفلاکس مزمن معده می باشد که باعث می شود سلول های پوششی آلوئولی صدمه ببینند و فرایند فیبروز را شروع کنند. این مسئله به خصوص در بیمارانی که دچار حملات فیبروز می شوند دیده شده است و تناسبی بین COMPILOBACTER در این حملات وجود دارد در مطالعات اخیر نشان داده شده است که داروهای درمان کننده ریفلاکس با کاهش مرگ و میر فیبروزی همراه می باشند.
نا توانی پوشش ریوی به دنبال صدمات وارد شده می تواند ایجاد فیبروز بکند این صدمات باعث می شود که صد وریدی آلوئول به هم بخورد و پروتئین های شامل فیبرین و فیبرونکتین وارد نسج ریه و آلوئول ها شود. در نهایت این باعث می شود که ساختمان ریه به هم بخورد و همراه با ناهنجاری بین فیبروساید ها و فیبروبلاست ها باعث فیبروز شود. شواهد زیادی وجود دارد که نشان می دهد فیبروبلاست ها منشا اصلی کلاژن در توسعه فیبروز می باشند. اما فیبروبلاست های مغز استخوان درصد کمی از سلول های مهاجر به ریه را تشکیل می دهند. تغییرات مزانشیالی به همراه اپیتلیالی که به آن EMT گفته می شود باعث تغییرات سلول های مزانشیالی می شود. گرچه این تغییرات در دوران نوزادی اهمیت دارد اما یک واکنش غیر معمول در مواجهه با صدمات می باشد. از طرفی دیگر TGFβ یک سایتوکاین مهم در شروع فرایند فیبروز می باشد.
درمان:
در سال 2005 اضافه کردن ACC به داروهای فیبروز توانست عملکرد ریه را تا حدودی نگه دارد به دنبال این مطالعه، مطالعه ی دیگری PANTHER انجام گرفت و نشان داد که بیمارانی که هم پردنیزولون هم آزاتیوپرین و هم ACC می خورند میزان مرگ و میر آنها بالاتر می روند به علاوه مطالعات بیشتر نشان داد که ACC به تنهایی نمی تواند موجب کاهش FVC در بیماران فیبروزی را بگیرد.در طول 5 سال گذشته دو دارو به نام پرفنیدون و نیمتدانیب برای بیماران فیبروزی معرفی شده است. در مطالعه ای به نام کپسیتی پرفنیدون به میزان 2400 میلی گرم در روز 1200 میلی گرم در روز برای 72 هفته داده شد. پرفنیدون در دوز 2400 میلی در روز توانست جلوی کاهش FVC را بگیرد. گرچه این یافته با مطالعه دیگر هم خوانی نداشت.
فاز سوم یک مطالعه به نام اسنت در آمریکا در حال انجام است. و نشان داده است که درمان با پرفنیدون باعث کاهش زیادی در بدتر شدن فیبروز کرده است و همچنین باعث جلوگیری از کاهش FVC بیش از 10 درصد شده است.تجمع دو مطالعه کپسیتی و اسنت نشان داد که پرفنیدون در دوز 2400 میلی گرم در روز موجب جلوگیری از کاهش FVC به مقدار بیش از 10 درصد و کاهش مرگ به میزان 43 درصد شد. به علاوه پرفنیدون موجب کاهش عوارض مرگ و میر در بیماران فیبروزی شد. داروی بعدی نینتدانیب می باشد. که یک مهار کننده تیروزینکیناز می باشد. این دارو موجب مهار فیبروبلاست ها از طرق مختلف مثل مهار VDGF, PDGF,FGF و در نهایت TGFβ می شود. مطالعه ای به نام تومارو پیشنهاد می کند که این دارو در دوز 150 میلی گرم دوبار در روز موجب جلوگیری از کاهش FVC می شود. به دنبال این مطالعه مطالعه دیگری به نام INPULSIS انجام گرفت و نشان داد که این دارو موجب کاهش افت FVC بعد از 52 هفته می شود. در مجموع این دارو به نظر می رسد موجب کاهش افت FVC به میزان 110 سی سی در سال می شود. که به مراتب کمتر از آنهایی هستند که این دارو را مصرف نمی کنند و کاهش FVC به میزان 5 تا 10 درصد دارند وقتی که دو مطالعه ی اخیر ترکیب شده اند بهبود مرگ و میر در این دارو مشاهده نشد. همچنین ترکیب این دو مطالعه نشان داد که این دارو میزان حملات فیبروز را کم می کند. مشابه پرفنیدون مطالعات نشان داد که این دارو در مراحل خفیف یا خیلی شدید به طور مساوی موثر هستند. مطالعه دیگری نشان داد که این داروها در یوآی پی اثبات شده و یو آی پی محتمل موثر می باشد. متاسفانه عوارض جانبی پرفنیدون نزدیک به 12 درصد و برای نینتدانیب 19 درصد می باشد. شایع ترین عوارض این داروها مربوط می شود به اسهال در 62 درصد و لک و پیس های حساس به نور که در 29 درصد گزارش شده است.
به علاوه هر دو دارو می توانند موجب افزایش آنزیم های کبدی بشوند.اما در واقعیت هر دو دارو در عمل به خوبی تحمل می شوند.
داروهای جدید:
MMP یک پروتئاز می باشد که در تغییر ساختمانی EMC نقش دارد. در این میان MMP7 شاخص ترین است و در بیماران فیبروزی بالا می رود و همراه است با شدت بیماری و کاهش TLCO و FVC به علاوه میزان ترکیبات از بین رفته توسط MMPA می تواند پیش آگهی بد IPF را پیش بینی کند همچنین سورفاکتانت SP-D و KL-6 که به موسین های نوع یک تلقی می شوند به میزان زیادی در ژاپن در طول 10 سال گذشته استفاده شده اند. افزایش هر دوی این مولکول ها همراه بوده است با بیماران فیبروزی اما هیچکدام نتوانسته اند موجب بهبود FVC – TLCO و بهبود مرگ و میری IPF شوند. با توجه به پیچیدگی بیماری فیبروز ممکن است در آینده داروهای جدیدی عرضه شوند. مثلا ژن سلول های منونوکلئار در 52 مورد همراه بوده است با پروگنوز فیبروز. تصحیح این ژن نیاز به مطالعات بیشتری دارد.
داروهای جدید روی مکانیزم های متفاوتی در درمان فیبروز اثر می گذارند و بسیاری از آنها در مراحل ابتدایی مطالعه هستند. یکی از راه های درمان فیبروز بر پایه ی این موضوع است که CTGF در بیماران سالم کم دیده می شود. اما در بیماران فیبروزی موجب افزایش TGFβ1 شده تولید ECM را زیاد می کند در حال حاضر یک مطالعه بر روی مهار کننده CTGF در فاز دوم به سر می برد. در مطالعه ای که روی موش ها انجام گرفت مهار TGFβ موجب شد که تخریب بافتی در فیبروز کاهش پیدا کند در حال حاضر این مطالعات در انسان در فاز یک به سر می برد.
اتوتوکسین یک آنزیمی است که در تولید LPA موثر است LPA در بیماران فیبروزی زیاد می شوند. یک مطالعه بر روی مهار کننده اتوتوکسین ها نشان داده است که میزان کاهش FVC را بعد از 12 هفته کاهش می دهد این مطالعه در فاز 3 قرار دارد. داروی دیگر مهار کننده IL13 است که در فاز دوم به سر می برد و بر اساس آخرین مطالعات این مهار کننده که بر علیه IL4 موثر بوده است در درمان فیبروز اثری نداشته است. داروی دیگر پنتراکسین 2 می باشد.که در واقع سرم آمیلوئید پی نامیده شود. میزان کم PTX/2 در بیماران فیبروزی کاهش پیدا کرده است. این دارو در فاز یک به سر می برد و توانسته است به خوبی توسط بیماران تحمل شود. داروی دیگر ژالکتین 3 می باشد که نقش مهمی در توسعه فیبروز از طریق فعالیت های ماکروفاژی و مایوفیبروبلاستی ایفا می کند. در حال حاضر مهار کننده این دارو در فاز اول به سر می برد.
با توجه به این که در 60 درصد از بیماران فیبروزی سلول های تی بر علیه کلاژن 5 وجود دارد. داروی دیگری ممکن است در این راه استفاده شود. فاز یک این دارو در حال انجام است.
دیدگاهها
خوراکخوان (آراساس) دیدگاههای این محتوا